Differentialdiagnose interstitieller Lungenerkrankungen
Akute Sarkoidose mit Löfgren-Syndrom und cutaner Sarkoidose im Tatoo - Acute Sarcoidosis (Löfgren-syndrome) with skin involvement in a tatoo
General Sherman's March to the Sea
Multilokuläre proximale Bronchialstenosen bei Sarkoidose – siehe Lungenfunktion
Bihiläre und mediastinale Sarkoidose nach Hoden-Carcinom – siehe Onkologie
Idiopathic Pulmonary Fibrosis IPF / Usual Interstitial Pneumonitis UIP (früh) (fortgeschritten) - English Abstract
Non-Specific Interstitial Pneumonitis NSIP - English Abstract
Respiratory Bronchiolitis Interstitial Lung Disease RB-ILD
Desquamative Interstitielle Pneumonitis DIP
Liquid-Aspiration bei einem Feuerschlucker
Docetaxel-induzierte interstitielle Pneumonitis mit Fibrose – siehe Onkologie
Sklerodermie (CREST-Syndrom) mit Lungenbeteiligung und pulmonaler Hypertonie
Pulmonale Lymphangioleiomyomatose LAM - English Abstract
Pulmonale Langerhans-Granulomatose (Histiozytosis X - HX)
Chronische Eosinophile Pneumonie CEP
Einteilung der Vaskuklitiden ANCA-assoziierte Vaskulitiden
Pulmonale Manifestation einer Wegenerschen Granulomatose WG
Mikroskopische Polyangiitis MPA mit langjährig vorausgehender interstitieller Lungenerkrankung
Churg-Strauss-Syndrom – siehe Allergologie
Idiopathische Lungenhämosiderose (M. Ceelen)
Reexpansionsödem nach Pneumothorax
Morbus Castleman - lokalisierter Typ
Morbus Castleman - multizentrischer Typ
In Bearbeitung befinden sich die folgenden Case Reports. Sie werden eingestellt, sobald ausreichend Platz für Bilder zur Verfügung steht.
Alveolar-Proteinose
Pulmonale Hypertonie bei Sklerodermie mit Lungenbeteiligung
Fibrosierende Lungenerkrankung bei Sklerodermie
Sekretgefüllte Bulla bei AT-1-Mangel
Schrotschußverletzung
Hämoptysen 55 Jahre nach Kriegsverletzung
Sarkoidose II mit Leberbeteiligung
Bronchiolitis Obliterans Organising Pneumonia BOOP / Cryptogen organisierende Pneumonie COP - infiltrativer Typ
Bronchiolitis Obliterans Organising Pneumonia BOOP / Cryptogen organisierende Pneumonie COP - medikamentös induziert
Bronchiektasen
Mediastinalemphysem
Zystische Bronchiektasen
Lungenembolie
Lungenembolie mit Infarktpneumonie
Sarkoid-Granulomatose
Gastric Banding
Aorteninsuffizienz
BOOP mit Einschmelzung
Alveoläre Hypoventilation
Stimmbandparese
Lymphangiosis carcinomatosa
Differentialdiagnose interstitieller Lungenerkrankungen
zum Inhaltsverzeichnis zur Start-Seite
Vor den einzelnen Fallbeschreibungen soll ein orientierender Überblick über die Differentialdiagnose der interstitiellen Lungenerkrankungen gegeben werden. Diese Übersicht soll helfen, sich in der Differentialdiagnose der interstitiellen Lungenerkrankungen zurecht zu finden. Sie soll kein Lehrbuch oder Literaturstudium ersetzen.
In der Differentialdiagnostik sind bei diffusen Lungenparenchymerkrankungen zunächst sekundäre Lungenerkrankungen (infolge Medikamentennebenwirkungen oder assoziiert mit Vaskulitiden), granulomatöse Lungenerkrankungen und einige andere abzugrenzen.
Diffuse Parenchymal Lung Disease DPLD (1)
Granulomatous DPLD
e.g. sarcoidosis
Idiopathic interstitial pneumonias
IIP
DPLD
of known cause
e.g. drugs,
collagen vascular disease
Other forms of DPLD
e.g. LAM, HX
Ein differentialdiagnostisches Konzept gibt das folgende flow-chart an (2). Das Procedere beschreibt folgendes flow-chart (2).
Bei den idiopathischen interstitiellen Pneumonien werden der idiopathischen pulmonalen Fibrose sechs weitere idiopathische interstitiellen Pneumonien gegenübergestellt, die sich in Hinblick auf radiologischen Befund, klinischen Verlauf und Therapie ansprechen unterscheiden.
Klinische und histologische Klassifikation der idiopathischen interstitiellen Pneumonien (1)
Klinisch / radiologisch / pathologisch
Histologie
Idiopathic pulmonary fibrosis IPF
Usual interstitial pneumonia UIP
Non-specific interstitial pneumonia NSIP
Non-specific interstitial pneumonia NSIP
Cryptogenic organising pneumonia COP
Organising pneumonia OP
Acute interstitial pneumonia AIP
Diffuse alveolar damage DAD
Respiratory bronciolitis interstitial lung disease RB-ILD
Respiratory bronchiolitis RB
Desquamative interstitial pneumonia DIP
Desquamative interstitial pneumonia DIP
Lymphocytic interstitial pneumonia LIP
Lymphocytic interstitial pneumonia LIP
Die histologischen Befunde werden wie folgt beschrieben.
Histologie der idiopathischen interstitiellen Pneumonie (1,2)
Zeitlicher Verlauf
heterogen
uniform
uniform
uniform
uniform
uniform
uniform
Interstitielle Entzündung
gering
meist deutlich
gering
gering
gering
gering
lympho
Kollagenfibrose
herdförmig
variabel, diffus
variabel, diffus
mild, fokal
nein
nein
nein
Fibroblasten-Proliferation
immer fibroblast foci
selten fibroblast foci
nein
nein
diffus
nein
nein
BOOP-Herde
nein
gelegent.
nein
nein
nein
ja
nein
wabiger Umbau
ja
selten
nein
nein
nein
nein
nein
Intraalveoläre Makrophagen
gelegent.
gelegent. herdförmig
diffus
peri-bronchio.
nein
schaumig
ja
Hyaline Membranen
nein
nein
nein
nein
selten
nein
nein
Die relative Häufigkeit der IIP in chirurgischen Biopsien variiert in den einzelnen Studien. Hier ist eine histologische Einteilung (3) nach einer älteren Klassifikation nach Katzenstein gezeigt, die DIP / RB-ILD zusammenfasste und AIP und LIP nicht anführte (3). Am häufigsten findet sich das histologische Bild einer UIP. Zu beachten ist auch die sicher unterrepräsentierte BOOP / COP. Der hochgradige klinische Verdacht einer BOOP / COP führt regelmäßig zu einer Therapie ohne histologische Sicherung.
Relative Häufgkeit der idiopathischen interstitiellen Pneumonien (3)
UIP
NSIP
DIP / RB-ILD
COP / BOOP
62 %
22 %
12 %
1%
In ihrem klinischen Verlauf unterscheiden sich die idiopathischen interstitiellen Pneumonien deutlich (3). Die NSIP wird in eine zellreiche und eine fibrotischen Form unterteilt. Die zellreiche Form zeichnet sich durch ein besseres Ansprechen auf Steroide und eine bessere Prognose aus.
Klinik der idiopathischen interstitiellen Pneumonien (2)
IPF
NSIP
DIP
RB-ILD
AIP
COP
LIP
Alter (Jahre)
65
55
40
35
49
55
45
Vorkommen bei Kindern
nein
ja
selten
nein
selten
ja
nein
Verlauf
chronisch
subakut / chronisch
chronisch
chronisch
akut
subakut
chronisch
Trommelschlegelfinger
häufig
selten
häufig
nein
nein
nein
selten
Fieber
selten
10–30 %
nein
nein
ja
ja
selten
Mortalität
70–90 %
11–60 %
0 – 27 %
0 %
62 %
gering
30 % ?
mittlere Überlebenszeit
2.8 J
4–13.5 J
12 J
nicht reduziert
1 –2 Mo
meist normal
70% normal
Ansprechen auf Steroide
schlecht
gut
meist
gut
schlecht
gut
meist gut
Im HR-CT zeigen sich zum Teil typische Befunde (1). Eine positive Diagnose einer UIP ist mittels HR-CT in 70 % möglich (4).
Radiologische Befunde der idiopathischen interstitiellen Pneumonien (1)
Diagnose
Verteilung
Verschattungsmuster
peripher, subpleural, basal
vorwiegend retikulär, Honeycombing, Traktionsbronchiektasen,
fokal Michglas (minimal)
diffus, peribronchial, basal, subpleurale Aussprarung möglich
vorwiegend Milchglas, retikulär, Traktionsbronchiektasen, alveoläre Konsolidierung (gering)
peripher, subpleural, basal
vorwiegend Milchglas,
retikulär gering,
geografisches Muster möglich
diffus
verdickte Bronchialwände, zentrilobuläre Knötchen, fleckförmig Milchglas
diffus
Milchglas mit fokaler Aussparung von Lobuli,
alveoläre Konsolidierung,
später Traktionsbronchiektasen
subpleural, peribronchial
vorwiegend fleckige alveoläre Konsolidierung,
mit / ohne Knötchen,
Milchglas (gering)
diffus, perivaskulär
Milchglas, perivaskuläre Zysten und Honeycombing, retikulär, Knötchen und Konsolidierung
(1) Travis WD, King TE, et al. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias . Am. J. Respir. Crit. Care Med. 165: 277-304. Free Full Text
(2) Katzenstein AL, Myers JL. Idiopathic Pulmonary Fibrosis. Clinical Relevance of Pathologic Classification. Am. J. Respir. Crit. Care Med. 157: 1301-1315. Free Full Text
(3) Flaherty KR, et al. Clinical significance of histological classification of idiopathic interstitial pneumonia Eur. Respir. J., Feb 2002; 19: 275 - 283. Abstract Free Full Text
(4) Johkoh T, et al. Idiopathic Interstitial Pneumonias: Diagnostic Accuracy of Thin-Section CT in 129 Patients Radiology 1999; 211: 555. Abstract Free Full Text
Dr. Holger Klee, 8 / 2007
zum Inhaltsverzeichnis zur Start-Seite
Pulmonale Lymphangioleiomyomatose LAM
zum Inhaltsverzeichnis zur Start-Seite
LAM is a very rare disease in young women, histological characterizised by a proliferation of pulmonary smooth muscles and lymph vessels. The lung is destroyed by thin-walled cysts. The patients suffer from dyspnoea. The lung function shows a bronchial obstruction.
Therapy consists of broncho dilatatory medication (beta adrenergic medication). In early cases a estrogen therapy may be an option.
Eine 52-jährige schlanke Patientin befand sich seit drei Jahren in regelmäßiger pneumologischer Behandlung wegen einer chronischen obstruktiven Atemwegserkrankung mit schwerer Überblähung. Es lag nur eine geringe Nikotinanamnese vor mit ca. 20 pack years. Alpha-1-Antitrypsin war normal.
Wegen rascher Progredienz und jetzt respiratorischer Partialinsuffizienz erfolgte ein diagnostisches Work-up.
Rö-Thorax HR-CT-Thorax
Konventionell-radiologisch stellt sich eine schwere Lungenüberblähung dar. Im HR-Thorax-CT fand sich überraschenderweise kein typisches Emphysem. Es liegt eine Zerstörung des Lungegewebes durch dünnwandige Zysten, sowie diskrete Milchglas-Trübungen. Das HR-CT-Bild ist für eine fortgeschrittene Lymphangioleiomyomatose typisch (1). Der makroskopische Befund entspricht dem HR-CT mit multiplen dünnwandigen Zysten. Histologisch finden sich ebenfalls dünnwandige, zum Teil subpleural gelegene Zysten mit Clustern von LAM-Zellen.
Bilaterale Zysten
100 %
Lineare Zeichnung
29 %
Milchglas-Trübung
12 %
Nodulär
11 %
Lymphadenopathie
9 %
Lungenfunktion
Die Lungenfunktion zeigte eine schwere bronchiale Obstruktion und schwere Überblähung. In der Blutgasanalyse spiegelt sich eine respiratorische Partialinsuffizienz wieder.
Lungenfunktion
VC
51.9 l
66 % des Solls
FEV-1
0.8 l
34 % des Solls
FEV1/VC
43 %
ITGV
4.3 l
167 % des Solls
R(tot)
0.63
pO2
59 mm Hg
ohne O2
pCO2
30 mm Hg
pO2
76 mm Hg
2 L O2/min
pCO2
32 mm Hg
Epidemiologie
Bei der pulmonalen Lymphangioleiomyomatose handelt es sich um eine sehr seltene Lungenerkrankung (ca. 1:1.000.000), die vorwiegend Frauen um das 35. Lebensjahr betrifft (2). Es kommt infolge einer somatischen Mutation zu einer nicht-malignen Proliferation von glatten Muskelzellen und Lymphgefäßen in der Lunge (3). Extrathorakal ist in 30 % ein Angiomyolipom der Nieren vorhanden. Spontan-Pneumothoraces kommen gehäuft vor.
Tuberöse Sklerose (M. Bourville-Pringle)
Eine Lymphangiomyomatose der Lunge kann auch im Rahmen einer Erbkrankheit vorkommen. Die tuberöse Sklerose (M. Bourneville-Pringle) ist mit einer Prävalenz von 1:10.000 deutlich häufiger und betrifft deutlich häufiger auch männliche Patienten. Hierbei sind extrathorakale Krankheitsmanifestationen häufiger (4).
Tuberöse Sklersoe (M. Bournville-Pringle)
Krampfanfälle
20 – 90 %
Mentale Retardierung
40 – 60 %
70 – 90 %
60 %
40 – 80 %
Cardiale Rhabdomyome
häufig
Unguale Fibrome
10 – 60 %
Pulmonale LAM
30 – 40 %
Klinik der LAM
Die Klinik der pulmonale LAM gleicht der eines schweren Emphysems mit Belastungsdyspnoe, Husten, aber auch Thoraxschmerz, Pneumothorax und Chylothorax. Die Lungenfunktion zeigt meist eine schwere Überblähung und bronchiale Obstruktion (1,5,6,7).
Diagnostik der LAM
Eine LAM sollte diferentialdiagnostisch bei typischer anamnestischer Konstellation in Betracht gezogen werden (junge Patientin mit schwerer Überblähung und nur geringer Nikotinanamnese, AT-1 normal). Das HR-CT-Thorax ist richtungsweisen. Die Diagnose kann über eine transbronchiale PE weiter gesichert werden.
Therapie der LAM
Zunächst erfolgt eine medikamentöse Therapie der bronchialen Obstruktion (5). LAM-Zellen können teilweise Östrogen- und Gestagenrezeptoren exprimieren. Daher sind Therapieversuche mit Medroxyprogesteron unternommen worden (6,7). Kontrollierte Studien hierzu liegen nicht vor. Der Spontanverlauf zeigt eine 10-Jahre-Prognose von ca. 80% (7,8). Die Prognose nach Lungen-Transplantation ist der anderer Indikationen vergleichbar (9,10).
Fallbericht
Bei unserer Patientin kam wegen weit fortgeschrittener Erkrankung kein Versuch einer Hormontherapie mehr in Frage. Die Patientin wurde erfolgreich einer Lungen-Transplantation zugeführt. Drei Jahre nach den Eingriff ist sie weiterhin gut belastbar. Die Lungenfunktion ist altersentsprechend normal.
Lungenfunktion nach Transplantation
VC
3.2 l
110 % des Solls
FEV-1
2.0 l
65 % des Solls
FEV1/VC
63 %
ITGV
3.1 l
119 % des Solls
R(tot)
0.13
pO2
95 mm Hg
ohne O2
pCO2
38 mm Hg
Wir verweisen noch abschließend auf den folgenden ausführlichen Case Report (11).
Literatur
(1) Avila NA, et al. (2002) Lymphangioleiomyomatosis: Correlation of Qualitative and Quantitative Thin-Section CT with Pulmonary Function Tests and Assessment of Dependance on Pleurodesis. Radiology; 223: 189.197. Free full text
(2) Johnson SR, Tattersfield AD (2000) Clincal experience of lymphangioleiomyomatosis in the UK. Thorax 55: 1052-1057. Abstract
(3) Atrinidis A, et al. (2000) Mutational analysis of the tuberous sclerosis gene TSC2 in patients with pulmonary lymphangioleiomyomatosis. J Med Genet 37: 55-57. Abstract
(4) Costabel U, Guzman J, Pulmonale Lymphangioleiomyomatose. In: Kirsten, Costabel Seltene Lungenkrankheiten 1, Großhansdorf: Inter-Pneu-Verlag, 2002: 3ff.
(5) Chu SC, et al. (1999) Comprehensive evaluation of 35 patients with lymphangioleiomyomatosis. Chest 115: 1041-1052. Free full text
(6) Kitaichi M, et al. (1995) Pulmonary lymphangioleiomyomatosis. A Report of 46 patients including clinopathologic study of prognostic factors. Am J Respir Crit Care Med 151: 527-533. Abstract
(7) Taylor WD, et al. (1990) Lymphangioleiomyomatosis. Clinical course in 32 patients. New Engl J Med 323: 1254-1260.
(8) Johnson SR (2006) Lymphangioleiomyomatosis. Eur Respir J; 27: 1056-1065. Abstract
(9) Trulick EP (1999) Lung transplantation: special considerations and outcome in LAM. In J Moss editor. LAM and other diseases characterised by smooth muscle proliertion.Marcel Dekker, New York, NY. 65 – 78.
(10) Boehler A, et al. (1996) Lung Transplantation for Leiomyomatosis. New Engl J Med 335: 1275-1280. Free full text
(11) Weir E, Cohen M. (2007) Teaching Case Report - A 48-year-old woman with lymphangioleiomyomatosis CMAJ 2007; 176 (9). Free Full Text
Dr. Holger Klee, 3 / 2007
zum Inhaltsverzeichnis zur Start-Seite
Pulmonale Langerhans-Granulomatose (Histiozytosis X)
zum Inhaltsverzeichnis zur Start-Seite
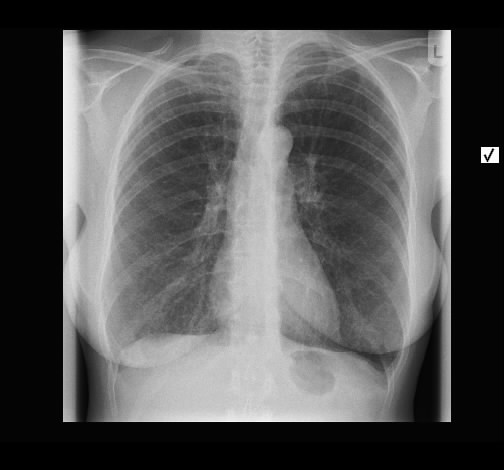
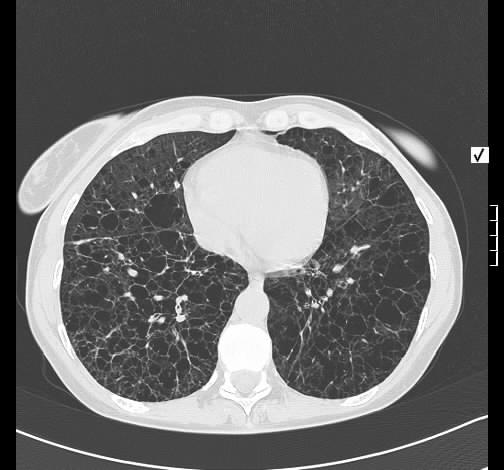
Wegen Polymyalgia rheumaticia war bei einer 60-jährigen Patientin die Einleitung einer systemischen Steroidtherapie geplant. Vor Therapieeinleitung wurde von der pulmonal beschwerdefreien Raucherin eine Röntgen-Thorax-Aufnahme angefertigt.
Hier zeigte sich eine ober- und mittelfeld-betonte feinnoduläre Zeichungsvermehrung, die sich im Thorax-CT wiederfindet. Angedeutet stellen sich keine Zysten mit breiter Wand dar.
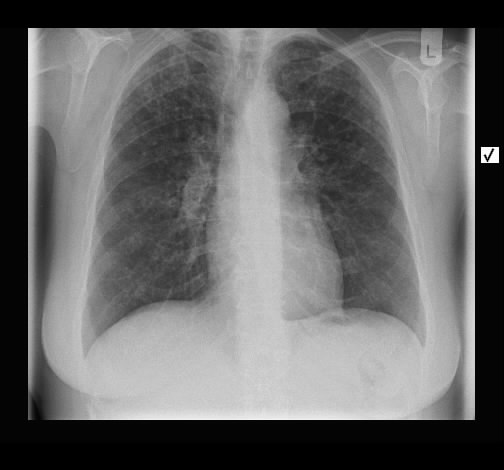
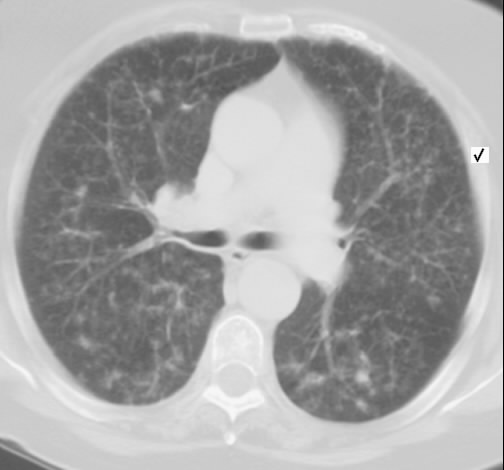
Rö-Thorax Thorax-CT
Diagnostik
Laboruntersuchungen, Mykobakteriologie und Lungenfunktion waren unauffällig. Eine Bronchoskopie mit peripherer PE waren ergebnislos. Eine BAL wurde nicht durchgeführt. Es finden sich typischerweise mehr als 3% CD1a-positive Zellen (6,9). Unter der Differentialdiagnose einer Lymphangiosis carcinomatosa wurde eine Tumorsuche durchgeführt ohne richtungsweisenden Befund. Eine mittels VATS (Video-assistierter Thorakoskopie) gewonnene PE (Histologie, Histologie 1,2,3) ergab die Diagnose einer pulmonalen Langerhans-Granulomatose (1,5).
Klinik der Langerhans-Granulomatosen
Bei der pulmonalen Langerhans-Granulomatose handelt es sich um eine eosinophile granulomatöse Erkrankung, die enge Beziehung zu der Abt-Letterer-Siwe-
und der Hand-Schüller-Christian-Erkrankung hat (5,8,10). Wobei diese Erkrankungen vorwiegend bei Kindern und Jugendlichen vorkommen. Pulmonale Histiozytosen sind selten mit einer Inzidenz von unter 1:100.000 (1,8,10).
Klinik der Langerhans-Granulomatosen
Pulm. Histiozytosis X
Abt-Letterer-Siwe
Hans-Schüller-Christian
Dyspnoe 90 %
Hepato-Splenomegalie
Landkartenschädel
Husten 70 %
Hautgranulomatose
Exophthalmus
Müdigkeit 30 %
Knochengranulomatose
Diabetes insipidus
Thoraxschmerz 20 %
Hämorrhagische Diathese
Kleinwuchs
Fieber 15 %
Radiologischer Befund und Verlauf der pHX
Typischerweise kommt es zu einer progredienten feinknotigen Infiltration mit Ausbildung von Zysten mit breiter Wand. Dies stellt sich entsprechend im HR-CT dar (1,2,4,6,7). Pneumothoraces können auftreten, wobei die Häufigkeit wahrscheinlich eher überschätzt wird und wohl bei etwa 5 % liegen dürfte (8). In fortgeschrittenen Stadien kommt es zu einer diffusen interstitiellen Gewebsvermehrung, die klinisch und radiologisch kaum von einem fortgeschrittenen Stadium der U.I.P. oder anderer interstitiellen Lungenerkrankungen unterschieden werden kann.
Therapie der pHX
Die pulmonale Histiozytosis X kommt nur bei Rauchern vor. Unter Nikotinkarenz kann eine Regredienz eintreten. Unter systemischen Steroiden ist ein rasches Ansprechen möglich (10).
Weiterer Verlauf des Fallberichtes
Aus pneumolgischer Sicht wurde zur Nikotinkarenz geraten. Eine Indikation zur systemischen Steroidtherapie ergab sich aus rheumatologischer Sicht ohnehin. Die Prognose wird als günstig angesehen mit einer mittleren Überlebenszeit von 13 Jahren (3,7,8,910).
Literatur
(1) Abbott GF, RadioGraphics 2004;24:821-841. Pulmonary Langerhans Cell Histiocytosis Free full text
(2) Brauner, et al. Pulmoanry Langerhans cell histiocytosis: evolution of lesions with high-resolution CT. Radiology 1997 (204) 497-502. Abstract pdf
(3) William CL, et al. Langerhans’-cell histiocytosis (histiocytosis X) a clonal proliferative disease. N Eng J Med 1994 (331) 154-160. Adstract
(4) Lacronique J, et al. Chest radiological features of pulmonary histiocytosis X: a report based on 50 adult cases. Thorax 1982 (37) 104-109. Abstract
(5) LCH-Register
(6) Moore ADA, et al. Pulmonary histiocytosis X: comparison of radiologic and CT findings. Radiology 1989 (172) 249-254. Abstract pdf
(6) Prasse A, Interstitielle Lungenerkankungen aufgrund bekannter Ursachen. In Matthys, Seeger: Klinische Pneumologie (2001) Springer, Berlin S. 401-3. BAL
(7) Schönfeld N, Clinical and radiologic features, lung function and therapeutic results in pulmonary histiocytosis X. Respiration 1993 (60) 38-44.
(8) Schönfeld N, Pulmonale Histiozytose X (Langerhans-Zell-Granulomatose). In Kirsten, Costabel: Seltene Lungenkrankheiten 1 (2002) Inter-Pneu-Verlag, Großhansdorf S. 41-74.
(9) Tazi A, Adult pulmonary Langerhans’ cell histiocytosis. Eur Respir J 2006; 27:1272-1285. Abstract
(10) Vassallo R, Pulmonary Langerhans’-cell histiocytosis. N Eng J Med 2000 (342) 1996-1978. Abstract
Dr. Holger Klee, 4 / 2007
zum Inhaltsverzeichnis zur Start-Seite
zum Inhaltsverzeichnis zur Start-Seite
Nach der Chapel-Hill-Klassifikation werden Vaskulitiden nach den betroffenen Gefäßen eingeteilt.
Chapel-Hill-Klassifikation der Vaskulitiden (1)
Giant-cell Arteriits
(Arteritis temporalis)
Granulomatöse Arteritis der Aorta und der großen Gefäße mit Prädilektion der extrakraniellen Äste der Carotis.
Oft A. temporails, meist Patienten über 50 Jahre und assoziiert mit Polymyalgia rheumatica
Takayasu-Arteritis
Granulomatöse Entzündung der Aorta und der großen Gefäße.
Meist Patienten jünger als 50 Jahre.
Polyarteritis nodosa
Nekrotisierende Entzündung von mitteleren und kleinen Arterien ohne Glomerulonephritis oder Vaskulitis der Arteriolen, Kapillaren oder Venen.
Kawasaki-Disease
Arteritis der großen, mittleren und kleinen Arterien, assoziiert mit einem mukokutanem Lymphknotensyndrom
Coronare Arterien häufig betroffen. Möglicherweise auch Aorta und Venen.
Meist Kinder
Wegener-Granulomatose
Granulomatöse Entzündung des Respirationstraks und nekrotisierende Vaskulitis der kleinen bis mittelgroßen Gefäße.
Häufig nekrotisierende Glumoerulonephritis.
Mikroskopische Polyangiitis
Nekrotisierende Vaskulitis der kleinen Gefäße mit geringen oder fehlenden Immun-Depositionen.
Möglicherweise Nekrotisierende Arteritis,
Sehr häufig nekrotosierende GN.
Häufig pulmonale Kapillaritis.
Churg-Struss-Syndrom
Eospinophilen-reiche und granulomatöse Entzündung des Respirationstrakts und nekrotisierende Vaskulitis der kleinen bis mittleren Gefäße, assoziiert mit Asthma und Eosinophlie.
Purupura Schönlein-Henoch
Vaskulitis der kleinen Gefäße mit IgA-Depositionen.
Typische Beteiligung von Haut, Magen-Darm-Trakt, Gelomeruli.
Assoziiert mit Arthralgie oder Arthritis
Essentielle Krypglobulinämie
Vaskulitis der kleinen Gefäße mit Kryoglubulin-Depositionen, assoziert mit Kryoglubulinen im Serum
Häufig Haut und Gloumeruli betroffen.
Leukozytoklastische Vaskulitis
Isolierte kutane leukozytoklastische Vaskulitis ohne systmische Vaskulitis oder Glomerulonephritis
Unter Berücksichtigung einer möglichen granulomatösen Reaktion ergibt sich die folgende Übersicht.
Klassifikation der primär systemischen Vaskulitiden (2,3)
Arteritis temporalis
Takayasu-Disease
Mittlere Gefäße
Polyareritis nodosa
Kawasaki-Disease
Kleine Gefäße
Wegener-Granulomatose
Mikroskopische Polyangiitis
Churg-Strauss-Syndrom
Purpura Schönlein-Henoch
Leukozytoklastische Vaskulitis
Essentielle Kyroglobulinämie
zum Inhaltsverzeichnis zur Start-Seite
Etwa 50% der systemischen Vaskulitiden sind ANCA-assoziiert. (3) Die Differentialdiagnose einer Vaskulitis mit Beteiligung des Respirationstrakts umfasst überwiegend die ANCA-positiven Vaskulitiden der kleinen Gefäße. (1,4) Dabei gibt die folgende Tabelle eine Übersicht über die klinische Präsentation.
Klinische Präsentation der ANCA-assoziierten Vaskulitiden (4,1)
M. Wegener
MPA
Churg-Strauss
Lunge
70 – 95
10 – 30
98 – 100
Obere Atemwege
70 – 95
0 – 15
20 – 70
Tracheobronchial
10 – 55
Niere
50 – 85
100
10 – 50
Haut
45 – 60
50 – 65
50 – 80
Muskuloskeletal
30 – 70
60
50
Augen
25 – 55
0 – 30
B-Symptome
15 – 45
70 – 80
70 – 80
Neurologisch
10 – 30
15 – 50
50 – 80
Herz
5 – 15
10 – 20
35 – 50
Gastrointestinal
30 – 45
30 – 60
Typische klinische Verläufe werden in den untenstehenden Case Reports dargestellt.
Die Wegenersche Granulomatose WG stellt ein oft fulminantes Krankheitsbild mit nekrotisch zerfallenden granulomatösen Lungenveränderungen und rapid progressiver Glomerulonephritis dar. Fast immer sind gegen Proteinase 3 (PR-3)-gerichtete c-ANCA nachweisbar.
Case Report: Pulmonale Manifestation einer Wegenerschen Granulomatose
Die mikroskopische Polyangiitis MPA manifestiert sich nach längerer Prodromalphase nahezu immer als Glomerulonephritis. Typisch ist eine pulmonale Vaskuklitis mit pulmonaler Hämorrhagie. Serologisch sind p-ANCA zu finden, die sich gegen Myeloperoxidase (MPO) richten.
Zytologie: c-ANCA und p-ANCA (4)
Röntgen und Histologie: Alveoläre Hämorrhagie (4)
Case Report: Mikroskopische Polyangiitis mit langjährig vorausgehender interstitieller Lungenerkrankung (wird in Kürze eingestellt)
Dem Churg-Strauss-Syndrom CSS geht in der Regel eine mehrjährige Asthma-Symptomatik voraus. Schließlich tritt eine systemische Klein-Gefäß-Vaskulitis mit eosinophilen pulmonalen Infiltraten, kardialen, gastrointestinalen und neurologischen (Mononeuritis multiplex) Beschwerden. Die klinische Symptomatik und die Prognose des CSS wird in der Hälfte der Fälle durch die kardiale Beteiligung bestimmt. p-ANCA sind häufiger als c-ANCA zu finden. (4)
Case Report: Churg-Strauss-Syndrom – siehe Allergologie
(1) Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med1997;337:1512–23.
https://content.nejm.org/cgi/content/full/337/21/1512
(2) Gross WL, Trabandt A, and Reinhold-Keller E. Diagnosis and evaluation of vasculitis. Rheumatology, March 1, 2000; 39(3): 245 - 252.
https://rheumatology.oxfordjournals.org/cgi/content/full/39/3/245
https://rheumatology.oxfordjournals.org/cgi/reprint/39/3/245
(3) Savage COS, Harper L, Adu D. Primary systemic vasculitis. Lancet1997;349:553–8.
https://download.thelancet.com/pdfs/journals/0140-6736/PIIS0140673697801183.pdf
(4) K. K. Brown. Pulmonary vasculitis. Proceedings of the ATS, January 1, 2006; 3(1): 48 - 57.
https://pats.atsjournals.org/cgi/reprint/3/1/48
Dr. Holger Klee, 4 / 2008
zum Inhaltsverzeichnis zur Start-Seite
Pulmonale Manifestation einer Wegenerschen Granulomatose WG
zum Inhaltsverzeichnis zur Start-Seite
Ein 53-jähriger Raucher wurde wegen Reizhusten ambulant pneumologisch vorgestellt. Es bestand nur eine geringe Beeinträchtigung des Allgemeinbefindens, keine Gewichtsabnahme, kein Nachtschweiß, keine Schmerzen, keine Hämoptysen. Entzündungsparameter waren nur gering erhöht.
Radiologisch fanden sich beidseits ausgedehntes, zum Teil nekrotisch zerfallendes Gewebe.
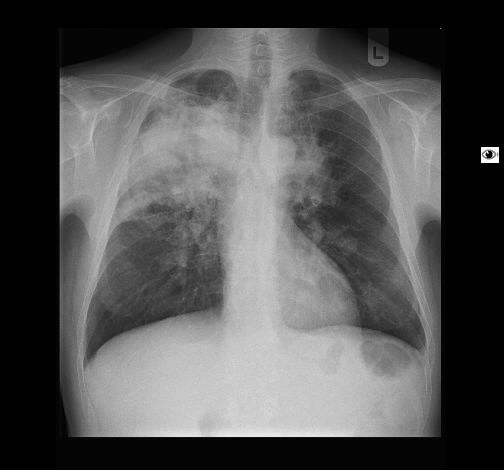
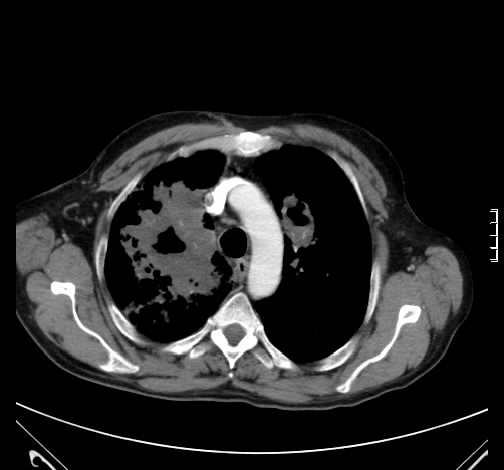
Rö-Thorax: Fremdgewebe re Oberlappen Thorax-CT: nekrotisch zerfallendes Fremdgewebe re Oberlappen
Endoskopisch stellt sich granulomatöses Gewebe dar, das makroskopisch neoplastischem Fremdgewebe nicht unähnlich ist.
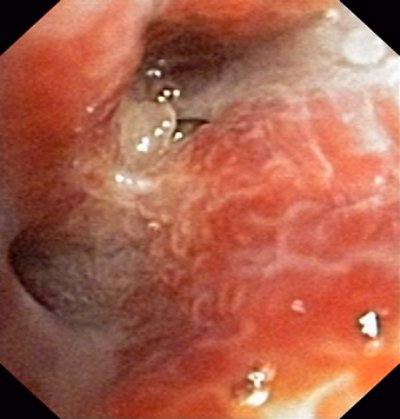
(Bronchoskopie)
Histologisch zeigte sich eine nekrotisierende Vaskulitis und eine pulmonale Kapillaritis. Makroskopisch stellen sich einschmelzende Nekrosen dar.
Histologie: nekrotisierende Kapillaritis (13)
Histologie nekrotisierende Vaskulitis pulmonale Kapillaritis
Histologie: Geographische Vaskulitis, Granulomatöse Entzündung (6)
multiple einschmelzende Nekrosen Pathologie
Im Labor fand sich c-ANCA und PR3-ANCA deutlich erhöht, so dass der hochgradige Verdacht auf eine pulmonale Manifestation eines M. Wegener erhoben werden mußte. Eine immunsuppressive Therapie wurde begonnen.
Klassifikation
Die Klassifikation der Vaskulitiden erfolgt nach den Empfehlungen der Chapel-Hill-Consensu-Konferenz nach dem histologisch sichtbaren Befall der Gefäße. Auf die Übersicht weiter oben sei verwiesen.
Einteilung der Vaskuklitiden (Chapel-Hill-Consensus) ANCA-assoziierte Vaskulitiden
Gefäßbefall bei Vaskulitiden (1)
Die Wegener’sche Granulomatose zählt zusammen mit der mikroskopischen Panarteritis und der Churg-Strauss-Vaskulitis zu den ANCA-positiven Vaskulitiden der kleinen Gefäße. Die Inzidenz der Wegener’schen Granulomatose wird mit 1 – 2 pro 100.00 angegeben. (1)
Praktisch regelhaft ist bei der Wegener’schen Granulomatose mit einer Beteiligung der oberen Atemwege zu rechnen. (1)
Organbeteiligungen bei Vaskulitiden (1)
Organbeteiligung bei M. Wegener (1,11)
Lunge
70 – 95 %
Obere Atemwege
70 – 95 %
Trachobronchial
10 – 55 %
Niere
50 – 85 %
Haut
45 – 60 %
Muskuloskeletal
30 – 70 %
Augen
25 – 55 %
B-Symptome
15 – 45 %
Neurologisch
10 – 30 %
Herz
5 – 15 %
Nicht alle initial begrenzte Erkrankungen müssen in einen gerneralisierte vaskulitischen Verlauf mit prognostisch sehr ungünstiger Nierenbeteiligung münden. Bei einer Gerneralisation können in der Regel PR3-ANCA nachgewiesen werden. (2) Der Spontanverlauf ohne Therapie ist dann in nahezu 100% letal. (1)
Therapie
Unter einer hochdosierten immunsuppressiven Therapie mit Steroiden und Cyclofosfamid kann in der überwiegend Mehrzahl eine Remission induziert werden. Rezidive treten in ca. 50% innerhalb von 5 Jahren auf. Rezidive sprechen meist gut auf eine erneute Immunsuppression an. (3) Längerfristige oder wiederholte immunsupressive Therapie kann mit erheblicher Morbidität und Mortalität einhergehen. So ist auch mit opportunistischen Infektionen wie der PCP zu rechnen. (4) Daher ist eine alternative immunsuppressive Therapie, z.B. mit Azathioprin wünschenswert. (5)
Es wird eine stadienabhängige Therapie empfohlen, die sich in Remissionsinduktion und Erhaltungstherapie, sowie ggf. Rezidivtherapie untergliedert. (6)
Die Induktionstherapie besteht aus einer Immunsuppression mit hocdosierten Steroiden und Cyclofosfamid. Bei leichterer Erkrankung kann ggf. Cyclofosfamid durch Methotrexat oder Azathioprin ersetzt werden, um ein günstigeres Toxizitätsprofil zu erreichen.
Schweregradeinteilung der ANCA-assoziierten Vaskulitiden und Therapie (6)
Allgemeinsym.
Nierenfunktion
Organbedroh.
Limited
Nein
Krea < 1.4
Nein
Steroide oder MTX oder AZA
Early generalized
Ja
Krea < 1.4
Nein
CYP + Steroide
MTX + Steroide
Active generalized
Ja
Krea < 5.7
Ja
CYP + Steoide
Severe
Ja
Krea > 5.7
Ja
CYP + Steroide + Plasmaaustausch
Refractory
Ja
Egal
Ja
Experimetelle Therapie ?
30 – 40 % der Patienten erleidet ein Rezidiv der Erkrankung. Ein erneutes Ansprechen auf die Induktionstherapie ist zu erwarten. Daher wird eine Erhaltungstherapie empfohlen. (6) Als Erhaltungtherapie eigenet sich Azathiprin in gleicher Weise wie Cyclofosfamid. Die Toxizität ist jedoch deutlich geringer. (7)
Erhaltungstherapie (7)
Steroide initial 1 mg/kg, reduziert bis 0.25 kg/kg
Cyclofosfamid 1.5 mg/kg /
Azathioprin 2 mg/kg
Nach 1 Jahr: Pred. 7.5 mg/d + AZA 1.5 mg/kg
Der Tumor-Nekosis-Faktor-Alpha-Inhibitor Etanercept hat sich als wirkungslos erwiesen. (8) Hauptproblem der Erhaltungstherapie ist die erhebliche Toxizität der Therapie und die langfristige Immunsuppression. (9)
In besonderen Situationen, wie therapierefraktärer systemischer Erkrankung oder therapierefraktärer diffuser alveolärer Hämorrhagie, wird eine Therapie mit Plasmaaustausch oder neueren Substanzen empfohlen. (6,7)
Mit dem Anti-CD20-Antikörper Rituximab, welcher in der Therapie maligner Lymphome etabliert ist, ist eine effektive Induktions- und Erhaltungstherapie der ANCA-assoziierten Vaskulitiden möglich.
Die Wirksamkeit von Mycofeonlat Mofetil und Leflumomide in der Erhaltungstherapie ist nicht abschließend geklärt. Bei refraktärer Vaskulitis ist ggf. der Einsatz von Anti-T-Zell-Globulinen zu erwägen. (7)
Therapie von Trachealstenosen
Bei tracheo-bronchialem Befall mit (typischerweise subglottischen) granulomatösen Stenosen wird eine Injektion von Steroiden empfohlen. Ggf. eine interventionelle Therapie mit endoskopischer Bougierung notwendig. (10)
Spirometrie bei subglottischer Stenose (12)
(1) Jennette JC, et al. Small-vessle vasculitis. New Eng J Med 1997 (337): 1512-1523 Extract Full Text
(2) Harrison DJ, et al. Antibodies to neurophil cytoplasmatic antigens in Wegeners granulomatosis and other conditions. Thorax 1989 (44): 373-377. Abstract
(3) Bacon PA The Spectrum of Wegeners Granulomatosis and Disease Relapse. New Eng J Med 2005 (352): 330-332 Abstract
(4) Ognibene FP, et al. Pneumocystis carinii pneumonia: a major complication of immunosuppressive therapy in patients with Wegeners granulomatosis. Am. J. Respir. Crit. Care Med., Vol 151, No. 3, Mar 1995, 795-799. Abstract
(5) Jayne D, et al. A Randomized Trial of Maintenance Therapy for Vaculitis Associated with Antineutrophil Cytoplasmatic Autoantibodies. New Eng J Med 2003 (349): 36-44. Abstract
(6) Frankel SK, et al. Update in the Diagnosis and Management of Pulmonary Vasculitis. Chest. 2006;129:452-465. PDF Free Full Text
(7) Jayner D, Rasmussen N, et al. A Randomized Trial of Maintenance Therapy for Vasculitis Associated with Antineutrophil Cytoplasmic Autoantibodies. N Engl J Med; 2003; 349:36. PDF
(8) WGET Research Group. Etanercept plus Standard Therapy for Wegener’s Granulomatosis. N Engl J Med; 2005: 352:351. PDF
(9) Langford CA. Treatment of ANCA-Associated Vasculitis. N Engl J Med; 2003: 349:3. PDF
(10) (3) K. K. Brown. Pulmonary vasculitis. Proceedings of the ATS, January 1, 2006; 3(1): 48 - 57. PDF
(11) Daum TE, et al. Tracheobronchial involvement in Wegeners granulomatosis. Am. J. Respir. Crit. Care Med., Vol 151, No. 2, 02 1995, 522-526. Abstract
(12) Thickett DR, etla. Pulmonary manifestations of anti-neutrophil cytoplasmic antibody (ANCA)-positive vasculitis. Rheumatology, March 1, 2006; 45(3): 261 - 268. Free Full Text
(13) Schwarz MI, Brown KK. Small vessel vasculitis of the lung. Thorax 2000;55:502–510. Free Full Text PDF
Dr. Holger Klee, 5 / 2007
zum Inhaltsverzeichnis zur Start-Seite
Mikroskopische Polyangiitis MPA mit langjähirg vorausgehender interstitieller Lungenerkrankung
zum Inhaltsverzeichnis zur Start-Seite
Seit ca. 3 Jahren war bei einem 71-jährigen Patieten eine nur langsma progrediente interstitielle Lungenerkrankung bekannt. Seit drei Wochen bestand eine Allgemeinverschlechterung, eine gering zunehmende Dyspnoe und subfebrile Temperaturen um 38.5° C.
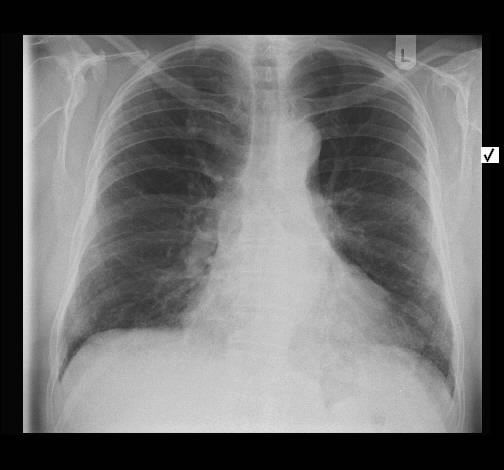
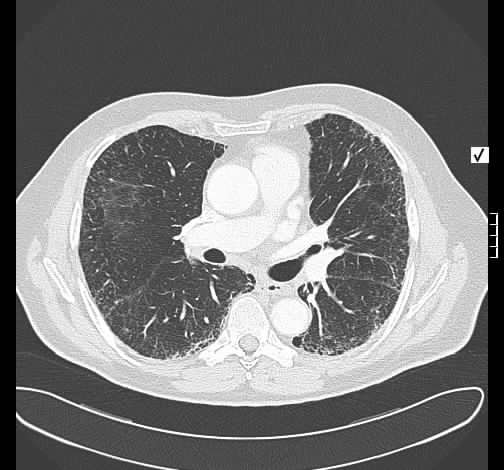
Rö-Thorax: retikuläre Zeichnungsvermehrung, kein frisches Infiltrat Thorax-CT: peripher betonte retikuläre Zeichnungsvermehrung
mit geringem honey-combing, kein Milchglas
Radiologisch stellte sich die vorbekannte interstitielle Lungenerkrankung ohne Änderung dar.
Im Labor fand sich eine mäßige CRP-Erhöhung und eine Mikrohämaturie. Die übrigen Laborparameter war nicht richtungsweisend.
Zunächst wurde unter dem Verdacht eines purulenten tiefen Atemwegsinfektes eine kalkulierte antibiostische Therapie eingeleitet. Zunächst mit Amoxicillin / Clavulansäure, später mit Ciprofloxacin, Piperacillin / Sulbactam, ohne Effekt auf die klinische Symptomatik. Der klinische Zustand verschlechterte sich langsam.
Unter weiterhin bestehenden Verdacht eines infektiologischen Krankheitsbildes wurde eine umfangreiche Diagnostik veranlasst, die sämtliche nicht richtungsweisend war. Die Differentialdiagnose einer Autoimmunerkrankung fühte zu weiterer serologischer Diagnostik. Es fanden sich p-ANCA mit einem Titer von 1:80 positiv und einer MPO-Ratio von 3.7. C-ANCA waren negativ.
Eine Phasenkontrastmikroskopie des Urins ergab Akanthozyten, war also eindeutig pathologisch. Die Nierenbiospie bestätigte eine fokal ausgebildete, floride nekrotisierende Glomerulonephritis, immunhistologisch ohne lineare oder granuläre Ablagerungen von IgA, IgG oder C3-Komplement.
Erythrozyten-Zylinder im Urin (12) Akanthozyten (16)
Im Verlauf trat eine retikuläre Hautefflureszenz an beiden Beinen auf. Eine Biopsie zeigte eine lympho-histiozytäre Vaskulitis.
Eine PE der klinisch unauffälligen Nasenschleimhaut zeigte eine granulomatöse Entzündung und perivaskulärer Fibrose, die mit einer Vaskulitis vereinbar war.
Somit wurde die Diagnose einer systemischen p-ANCA-positiven Vaskulitis (mikroskopischen Polyangiitis) mit Nieren- und Hautbeteiligung gestellt. Möglicherweise war die vorbekannte interstitielle Lungenerkrankung ebenfalls im Rahmen der Vaskulitis zu sehen.
Es wurde eine Immunsuppression mit Cyclofosfamid und Prednisolon eingeleitet. Hierunter konnte eine rasche Besserung des Allgemeinzustandes erreicht werden. Die Temperaturen war innerhalb eines Tages rückläufig.
Nach drei Wochen waren p-ANCA auf die Hälfte gefallen und der Phasenkontrastbefund des Urins unauffällig. Kreatinin fiel von 1.6 auf 1.3 mg/dl ab.
Die mikroskopische Polyangiitis MPA zählt zu den ANCA-assoziierten Angiitiden der kleinen Gefäße.
Einteilung der Vaskuklitiden (Chapel-Hill-Consensus) ANCA-assoziierte Vaskulitiden
Die Indzidenz der Wegener’schen Granulomatose wird in den USA mit ca. 2 pro 100.000 angegeben. (1,2) Die MPA ist in etwa 5-mal seltener, so dass eine Inzidenz von ca. 0.5 pro 100.000 angenommen werden kann.
Der Manifestation einer systemischen Vaskulitis geht bei der MPA in der Regel ein längeres Prodromalstadium mit Allgemeinverschlechterung, subfebrilen Temperaturen, Gewichtsabnahme oder Arthralgien voraus. Das Auftreten einer Glomerulonephritis ist nahezu obligat. (3)
Organbeteiligung bei MPA (3)
Rapid progressive GN
100 %
Lunge (Hämorrhagie/-ptysis)
10 - 30 %
B-Symptome
70 – 80 %
Haut
60 – 65 %
Nervensystem
15 – 50 %
Gastrointestinal
30 – 45 %
Augen
0 – 30 %
Herz
10 – 20 %
Obere Atemwege
0 – 15 %
Die häufigste pulmonale Manifestation ist eine diffuse alveoläre Hämorrhagie. (3) Die Differentialdiagnose der pulmonale Kapillaritis umfasst jedoch ein Reihe weiterer Ekrankungen und auch Medikamentennebenwirkungen. (4,5)
Differentialdiagnose der pulm. Kapillaritis (4)
Antiphospholipid-Syndrom
Behcet-Syndrom
Diphenylhydantoin
Goodpasture-Syndrom
Purpura Schönlein-Henoch
Idiopathisches pulmo-renales Syndrom
IgA-Nephritis
Mikroskopische Polyangiitis
Systemischer Lupus erythematosus
Wegener’sche Granulomatose
Histologie: Pulmonale Hämorrhagie (1) Histologie: alveoläre Hämorrhagie bei nekrotisierender Kapillaritis (9)
Rö + Histologie: Alveoläre Hämorhagie (3) Histologie: frühe pulmonale Kapillaritis ohne Nekrosen oder Hämorrhagie (9)
Eine pulmonale Fibrose kann der systemischen Vaskulitis um Jahre vorausgehen. (6,7) Auch ungewöhnliche Primär-Manifestationen sind möglich, wie mediastinale Raumforderungen. (8)
Labor
Es lassen sich häufiger p-ANCA, die gegen Myeloperoxidas (MPO) gerichtet sind nachweisen, als c-ANCA. (1,2,3,9,10)
Trotz hoher Sensitivität (99 %) und guter Spezifität (70 %), ist der Nachweis von ANCA nicht als Beweis für das vorliegen einer primären systemischen Vaskulitis anzusehen. Insbesondere p-ANCA / MPO-ANCA treten in Assoziation zu malignen Lymphomen auf. (11)
Wegen der Seltenheit der Erkrankung existieren keine eigenständigen Therapiestudien zur MPA. Vorliegende Untersuchungen fassen die MPA mit der Wegener’schen Granulomatose zusammen. Es wird daher eine stadienabhängige Therapie in Anlehnung an die Wegener’sche Granulomatose empfohlen, die sich in Remissionsinduktion und Erhaltungstherapie, sowie ggf. Rezidivtherapie untergliedert. (12)
Die Induktionstherapie besteht aus einer Immunsuppression mit hocdosierten Steroiden und Cyclofosfamid. Bei leichterer Erkrankung kann ggf. Cyclofosfamid durch Methotrexat oder Azathioprin ersetzt werden, um ein günstigeres Toxizitätsprofil zu erreichen.
Schweregradeinteilung der ANCA-assoziierten Vaskulitiden und Therapie (12)
Allgemeinsym.
Nierenfunktion
Organbedroh.
Limited
Nein
Krea < 1.4
Nein
Steroide oder MTX oder AZA
Early generalized
Ja
Krea < 1.4
Nein
CYP + Steroide
MTX + Steroide
Active generalized
Ja
Krea < 5.7
Ja
CYP + Steoide
Severe
Ja
Krea > 5.7
Ja
CYP + Steroide + Plasmaaustausch
Refractory
Ja
Egal
Ja
Experimetelle Therapie ?
30 – 40 % der Patienten erleidet ein Rezidiv der Erkrankung. Ein erneutes Ansprechen auf die Induktionstherapie ist zu erwarten. Daher wird eine Erhaltungstherapie empfohlen. (1,2,12) Als Erhaltungtherapie eigenet sich Azathiprin in gleicher Weise wie Cyclofosfamid. Die Toxizität ist jedoch deutlich geringer. (CYCAZAREM-Trial) (13)
Erhaltungstherapie (13)
Steroide initial 1 mg/kg, reduziert bis 0.25 kg/kg
Cyclofosfamid 1.5 mg/kg
Azathioprin 2 mg/kg
Nach 1 Jahr: Pred. 7.5 mg/d + AZA 1.5 mg/kg
Der Tumor-Nekosis-Faktor-Alpha-Inhibitor Etanercept hat sich als wirkungslos erwiesen. (14) Hauptproblem der Erhaltungstherapie ist die erhebliche Toxizität der Therapie und die langfristige Immunsuppression. (15) In einer neueren Studie (WYGENT-Trial) fand sich kein signifikanter Unterschied zwischen Azathioprin und Methotrexat in der Rezidivprophylaxe. Es bestand jedoch ein nicht-signifikanter Trend zu schlechterer Verträglichkeit und geringer Wirksamkeit von MTX. (17)
In besonderen Situationen, wie therapierefraktärer systemischer Erkrankung oder therapierefraktärer diffuser alveolärer Hämorrhagie, wird eine Therapie mit Plasmaaustausch oder neueren Substanzen empfohlen. (10,12)
Mit dem Anti-CD20-Antikörper Rituximab, welcher in der Therapie maligner Lymphome etabliert ist, ist eine effektive Induktions- und Erhaltungstherapie der ANCA-assoziierten Vaskulitiden möglich.
Die Wirksamkeit von Mycofeonlat Mofetil und Leflumomide in der Erhaltungstherapie ist nicht abschließend geklärt. Bei refraktärer Vaskulitis ist ggf. der Einsatz von Anti-T-Zell-Globulinen zu erwägen. (12)
(1) Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med1997;337:1512–23. Free Full Text
(2) Savage COS, Harper L, Adu D. Primary systemic vasculitis. Lancet1997;349:553–8. PDF
(3) K. K. Brown. Pulmonary vasculitis. Proceedings of the ATS, January 1, 2006; 3(1): 48 - 57. PDF
(4) Green RJ, et al. Pulmonary Capillaritis and Alveolar Haemorrhage. Update on Diagnosis and Management. Chest 1996; 110: 1305-16. PDF
(5) Gross WL, Trabandt A, and Reinhold-Keller E. Diagnosis and evaluation of vasculitis. Rheumatology, March 1, 2000; 39(3): 245 - 252. Free Full Text PDF
(6) Eschun GM, Steven N. Mink, and Sat Sharma. Pulmonary Interstitial Fibrosis as a Presenting Manifestation in Perinuclear Antineutrophilic Cytoplasmic Antibody Microscopic Polyangiitis. Chest, Jan 2003; 123: 297 - 301. PDF
(7) Eschun GM, et al. Fibrosing Alveolitis is often Associated With Microscopic Polyangiits and P-Antineutrophil Cytoplasmatic Antibody (P-ANCA). Chest (114, Supplement); 1998: 347S. PDF
(8) Fidder HH, et al. Hemoptysis and a Mediastinal Mass. Chest. 1999;115:1473-1475. PDF Free Full Text
(9) Schwarz MI, Brown KK. Small vessel vasculitis of the lung. Thorax 2000;55:502–510. Free Full Text PDF
(10) Thickett DR, etla. Pulmonary manifestations of anti-neutrophil cytoplasmic antibody (ANCA)-positive vasculitis. Rheumatology, March 1, 2006; 45(3): 261 - 268. Free Full Text
(12) Frankel SK, et al. Update in the Diagnosis and Management of Pulmonary Vasculitis. Chest. 2006;129:452-465. PDF Free Full Text
(13) Jayner D, Rasmussen N, et al. A Randomized Trial of Maintenance Therapy for Vasculitis Associated with Antineutrophil Cytoplasmic Autoantibodies. N Engl J Med; 2003; 349:36. PDF
(14) WGET Research Group. Etanercept plus Standard Therapy for Wegener’s Granulomatosis. N Engl J Med; 2005: 352:351. PDF
(15) Langford CA. Treatment of ANCA-Associated Vasculitis. N Engl J Med; 2003: 349:3. PDF
(16) Maslak, P. Akanthozytosis. Amaerican Society of Hemotology, image bank; 2005: 0918: 101405. Free Full Text
(17) Pagnoux C, et al. Azathioprine or Methotrexate Maintenance for ANCA-Associated Vasculitis. NEJM; 2008: 359: 2790. Abstract Full Text
(18) Klee H, Schaberg T, Fibrosierende Lungenerkrankung drei Jahre vor systemischer Vaskulitis bei Mikroskopischer Polyangiitis, Posterpräsentation auf dem Kongress der Deutschen Gesellschaft für Pneumologie und Beatmungsmedizin 2009. Abstract
Dr. Holger Klee, 5 / 2008
zum Inhaltsverzeichnis zur Start-Seite
Idiopathic Pulmonary Fibrosis IPF / Usual Interstitial Pneumonia UIP (früh) (fortgeschritten)
zum Inhaltsverzeichnis zur Start-Seite
IPF/UIP is the most common interstitial lung disease. Cinically it impresses as progressive dyspnoea and unproductive cough. Sclerosiphony may be present. In lung function restriction and diminished diffusion capacity are seen.
The main finding in radiological examination is an unequal distribution of fibrotic pattern. The disease progresses timely and regionally heterogen. So one can find severely fibrotic parts and traction bronchiectases adjected to normal lung tissue. Fibrotic changes predominate in peripher / subpleural and basal parts of the lung. Subpleural so called „honey combing“ is a typical, if not pathognominic finding. Small ground glass opacities may be present non the less.
The prognosis remains unfavorable. (50% in 3 years) There is no established therapy for IPF/UIP. In young patients not older than 60 years of age, the diagnosis should be established by thoracoscopy and a bilateral lung-transplantation should be considered.
Ein 52-jähriger Patient stellt sich wegen chronischem unproduktivem Reizhusten und geringer Belastungysdyspnoe vor. Es bestanden kein Fieber oder Thoraxschmerzen. Die weitere Anamnese war unauffällig ohne Hinweis auf eine rheumatologische Erkrankung, keine Haustiere, keine berufliche Exposition zu inhalativen Allergenen oder Noxen. Der Patient nahm keine regelmäßige Medikamention ein.
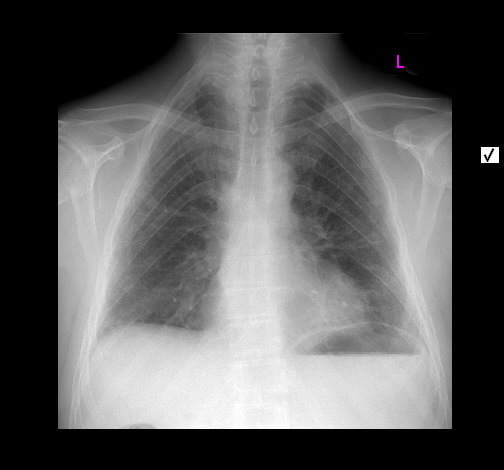
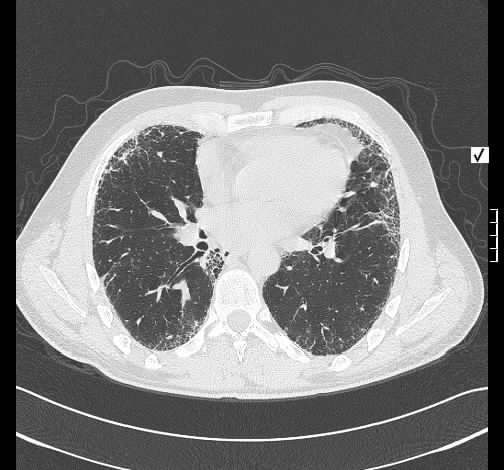
Rö-Thorax: diskrete periphere retikuläre Zeichungsvermehrung HR-CT: diskontinuierlich verteilte periphere reikuläre Zeichnung
beginnendem Honey combing, kein Milchglas
Die radiologischen Befunde waren gut mit einer beginnenden idiopathischen pulmonalen Fibrose vereinbar.
Lungenfunktion
Lungenfunktion
VC
5.0 l
98 % des Solls
FEV-1
3.9 l
78 % des Solls
ITGV
2.9 l
99 % des Solls
R(tot)
0.11
TLCO/VA
87 % des Solls
pO2
74 mm Hg
pCO2
40 mmHg
In der Lungenfunktion war keine Restriktion oder Diffusionsstörung zu sehen.
Es erfolgte eine differentialdiagnostische Klärung. Bronchoskopie mit BAL, peripherer PE waren die nicht richtungsweisend. Es schloß sich eine video-assistierte Thorakoskopie VATS mit Lungen-PE an. Hier wurde die histologische Diagnose einer UIP gestellt.
Klassifikation
Nach der Klassifikation der Consensus-Konferenz der ATS und ERS (1) handelt es sich bei der IPF um eine idiopathische interstitielle Lungenerkrankung mit klinischen, radiologischen und histologischen Chrakteristika und schlechtem Therapieansprechen und daher schlechter Prognose. (2,3)
Differentialdiagnose interstitieller Lungenerkrankungen (1) Diagnostischer Prozess bei ILD
Differentialdiagnose interstitieller Lungenerkrankungen (Übersicht weiter oben)
Die Datenlage zur Epidemiologie ist spärlich. Die Indizenz ist regional wahrscheinlich unterschiedlich. Mit einer für Deutschland geschätzten Prävalenz von 6/100.000 ist die IPF nach der Sarkoidose mit 10/100.000 die zweithäufigste idiopathische interstitielle Lungenerkrankung. Eine amerikanische Studie gibt eine Prävalenz von 13 - 20/100.000 an. (15) Das Erkrankungsalter liegt meist über 60 Jahre. Männer sind etwas häufiger betroffen. Rauchen gilt als Risikofaktor. (1,2,3,4,5)
Pathogenese
Die Pathogenese der IPF / UIP ist nicht geklärt. Es finden sich zwar regelhaft entzündliche Veränderungen. Diese sind jedoch nicht als pathogenetisch wirksam anzusehen, sondern eher ein Epiphänomen. Primär kommt es bei entsprechender genetischer Disposition unter Einfluß eines nicht näher bekannten Triggers zu einer Störung der Integrität der alveolären Basalmembran. Bei der frustranen Regeneration kommt es zur Proliferation von Alveolarzellen II. Mesenchymzellen wandern in den Alveolarraum und führen zu Exsudation und Fibrosierung. Unter dem stimulierenden Einfluß von TGF-ß (transforming growth factor ß) wird vermehrt extrazelluläre Matrix gebidet. (1,2,3,4)
Klinisch steht zunächst insbesondere eine Belastungsdyspnoe im Vordergrund, die innerhalb weniger Wochen bis Monate deutlich zunimmt. Weiterhin beklagen manche Patienten einen unproduktiven Hustenreiz. Später entwickeln sich Folgen der chronischen Hypoxämie wie Rechtsherzbelastung, Uhrglasnägel und Trommelschlegelfinger. Bei der klinischen Untersuchung imponieren Tachypnoe, hochstehend, kaum verschiebliche Lungengrenzen und basales Knisterrasseln (Sklerosiphonie). (1,2,4)
Diagnostik
Im Rahmen der Diagnostik sollte eine Diagnosesicherung angestrebt werden und die aktuelle funktionelle Einschränkung dokumentiert werden. (2)
Gold-Standard der Differentialdiagnose ist zwar eine chirurgische Lungebiopsie mittel VATS. Doch ist dies einer Vielzahl von Patienten funktionell kaum zumutbar. Nach den Empfehlung der ATS und ERS kann unter bestimmen Voraussetzungen zur Diagnosesicherung auf eine chirurgische Biospie verzichtet werden. Insbesondere sollte alternative Diagnosen weitgehend ausgeschlossen werden. Hierzu können eine gezielt erweiterete Anamnese, Laboruntersuchungen, Broncho-alveoläre Lavage und transbronchiale Lungenbiopsie dienen.
Diagnose einer Idiopathischen Pulmonlen Fibrose IPF (1,2)
Hauptkriterien
Ausschluß alternativer Diagnose
Restriktion und Diffusionsstörung
Retikuläre Zeichnungsvermehrung bds. bas. allenfalls geringes Milchglas
BAL und TBB ohne Hinweis für alternative Diagnose
Nebenkriterien
Alter > 50 Jahre
Plötzlich einsetzende Belastungsdyspnoe
Krankheitsverlauf > 3 Monate
Sklerosiphonie
Gesicherte Diagnose bei allen Hauptkriterien und mindestens 3 Nebenkriterien
Lungenfunktion
Lungenfunktionell besteht eine restriktive Funktionseinschränkung und Diffusionsstörung. Oxygerometrisch / spiroergometrisch kann einen Belastungshypoxämie mit hoher AaDO2 als sehr sensitiver Parameter erfaßt werden. (2,4,6)Als einfacher – prognostisch verwertbarer – Parameter zur Verlaufsbeurteilung kann der 6-Minuter-Gehtest herangezogenwerden. (7,8)
Radiologische Befunde
Im Röntgen-Thorax imponiert eine basal und peripher betonte retikuläre Zeichnungsvermehrung, die später zur Unterlappenschrumpfung und evtl. zur Verziehung führt. Im HR-CT stellt typischerweise eine diskontinuierliche Verteilung der Veränderungen dar. Neben Arealen mit retikulärem Muster, das in wabigem-kleinzystischen Umbau (Honey combing) übergeht, finden sich nahezu unbeteiligte Lungenabschnitte. Die pathologischen Veränderungen betreffen bevorzugt periphere und basale Lungenabschnitte. Weiter typische Befunde sind Traktions-Bronchiektasen und Pleuraverdickungen. Milchglasverschattungen können vorkommen. Sie bilden jedoch nicht das vorherrschende Merkmal. (1,2,4,9,10)
Typische radiologische Befunde bei IPF (1)
Verteilung
Muster
Peripher
Retikulär
Subpleural
Honeycombing
Basal
Traktionsbronchiektasen
Fokal minimal Milchglas
Typische Radiologische Befunde verschiedener Interstitieller Lungenerkrankungen (1)
Histologie
Histologisch findet sich die oben beschriebene diskontinuierliche Verteilung der pathologischen Veränderungen wider. Neben Zonen mit Fibroblastenfoci und Verbreiterung des Interstitiums und möglicherweise Narbenbildungen finden sich Bereiche mit normalem Lungengewebe. (1,3,4,5,11) Die Befunde der BAL sind unspezifisch. Es zeigt sich eine Neutrophilie, evtl. eine diskrete Eosinophilie. (1,2,4,12)
Typische histologische Befunde bei IPF (1)
Zeitlicher Verlauf
Interstitielle Entzündung
Gering
Kollagenfibrose
Fibroblasten-Proliferation
Immer
BOOP-Herde
Nein
Wabiger Umbau
Ja
Intraalveoläre Makrophagen
gelegentlich
Hyaline Membranen
Nein
Histologie - Übersicht Histologie - Fibroblastenfokus Histologie - Übersicht
Histologie - Übersicht Histologie - Fibroblastenfokus Histologie - Fibroblastenfokus
Therapie
Obwohl pathogenetisch kein entzündlicher Prozeß im Vordergrund steht und die Wirksamkeit entsprechend begrenzt ist, wird eine antientzündliche / immunsuppressive Therapie empfohlen. (2,4)
Zunächst kommt eine systemische Steroidtherapie in Frage mit 0.5 mg/kg Prednisolon-Äquivalent in fallender Dosierung über mehrere Wochen. Weiterhin sollte die Therapie ergänzt werden um ein Immunsuppressivum, beginnend mit Azathioprin 2-3 mg/kg (bis 150 mg). Bei Erfolglosigkeit kann Cyclofosfamid 2 mg/kg (einschleichend bis 150 mg)gegeben werden. Eine Kombination mit ACC 3 x 600 mg als sogenannte „Tripple-Therapy“ kann möglicherweise die Prognose etwas verbessern. (2,4,13) Interferon Gamma ist zurzeit noch keine etablierte Therapieoption (14).
Als Zeichen des Therapieversagens gilt eine Veschlechterung der statischen Lungenvolumina VC bzw. TLC um 10 % oder der Diffusion um 15 %. (2,4)
Palliativ sollte bei zunehmender respiratorischer Insuffizienz eine Sauerstofflangzeittherapie eingeleitet werden. Hierbei kann man sich an der Richtlinien der Deutschen Gesellschaft für Pneumologie und Beatmungsmedizin orientieren. (2,4)
Prognose
Die Prognose ist auch unter der o.a. Medikation schecht. Die mittlere Überlebenszeit beträgt 3 Jahre. Daher sollte bei Diagnosestellung zügig die Möglichkeiten einer Transplantation geklärt werden. Da das Manifestationsalter der Erkrankung meist über 60 Jahre liegt, ist eine Transplantation für diese älteren Patienten keine Option. (2,4,13)
Prognose-Unterschiede UIP - NSIP - RB-ILD/DIP
Idiopathic Pulmonary Fibrosis IPF / Usual Interstitial Pneumonia UIP (fortgeschritten) (früh)
zum Inhaltsverzeichnis zur Start-Seite
Bei histologisch gesicherter UIP wurde eine systemische Steroidtherapie begonnen. Hierunter konnte kein Ansprechen erreicht werden. Daher wurde eine immunsuppressive Therapie mit Azathioprin eingeleitet. Diese mußte wegen Unverträglichkeit abgebrochen werden und wurde durch Cyclofosfamid ersetzt. Auch hierunter war ein weiterer Krankheitsprogress nicht zu verhindern.
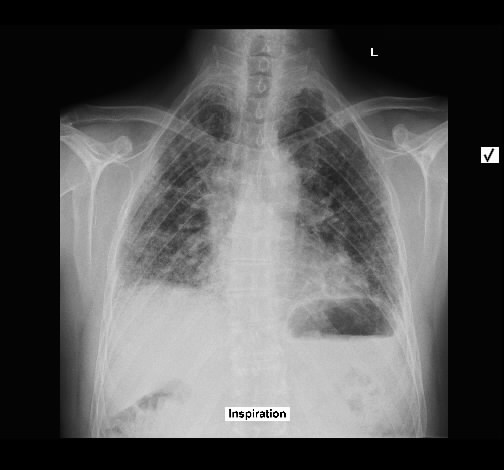
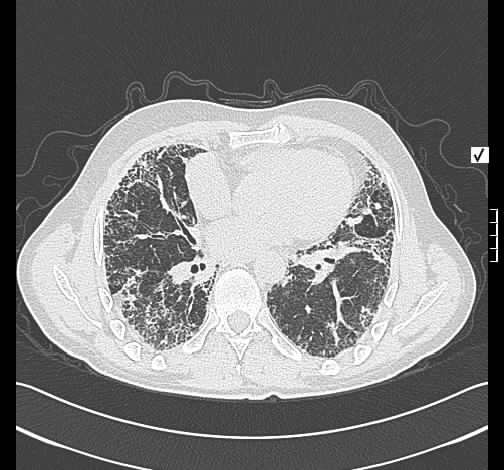
Rö-Thorax: ausged. retikuläre Zeichnungsvermehrung mit UL-Schrumpfung HR-CT: diskontinuierliche retikuläre Zeichnungsvermehrung
mit Honey combing und Traktionsbronchiektasen
Pleuraverbreiterung
Nach drei Jahren bestand radiologisch ein IPF-typisches Bild.
Lungenfunktion
Lungenfunktion
VC
1.5 l
34 % des Solls
FEV-1
1.2 l
36 % des Solls
ITGV
2.6 l
94 % des Solls
R(tot)
0.18
TLCO/VA
54 % des Solls
pO2
64 mm Hg
pCO2
40 mmHg
Funktionell war es zu einer erheblichen Einschränkung gekommen mit schwerer Restriktion, Diffusionsstörung und grenzwertiger Hypoxämie in Ruhe und schwerer Belastungshypoxämie.
Spiroergometrie
Spiroergemetrisch bestehen Zeichen einer schweren Diffusions- und Verteilungsstörung mit schwerer belastungsinduzierbarer Hypoxämie spirographisch Hinweise auf eine restriktive Störung.
Spiroergometrie
3 / 2002
2 / 2005
VO2max. (%)
87
57
VO2/kg
27
14
Last (W)
196
78
PO2min. (mmHg)
52
38
AaDO2 (mmHg)
58
64
VE/VO2
35
45
PETCO2 (mmHg)
50
30
Nach Ausschöpfung aller medikamentöser Therapiemöglichkeiten wurde der Patient zur bilateralen Transplantation vorgestellt. Diese wurde komplikationslos durchgeführt.
Literatur
(1) ATS & ERS. American Thoracic Society / European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am J RespirCrit Care Med (2001); 165, 277-304. Free Full Text
(2) ATS & ERS. Idiopathic Pulmonary Fibrosis: Diagnosis and Treatment.International ConsensusStatement.Am J Respir Crit Care Med(2000); 161, 646-664. Free Full Text
(3) Katzenstein, AL, Myers JL.Idiopathic PulomonaryFibrosis. Clinical Relevance of Pathologic Classification. Am J Respir Crit CareMed (1998); 157: 1301-1315. Free Full Text
(4) Bewig, B. Idiopathische Lungenfibrose, Der Pneumologe (2005) 2; 457-468.
(5) G. Raghu, D. Weycker, J. Edelsberg, W. Z. Bradford, and G. Oster Incidence and Prevalence of Idiopathic Pulmonary Fibrosis Am. J. Respir. Crit. Care Med., October 1, 2006; 174(7): 810 - 816. Abstract
(6) F. J. Martinez and K. Flaherty. Pulmonary function testing in idiopathic interstitial pneumonias. Proceedings of the ATS, January 1, 2006; 3(4): 315 - 321. Abstract
(7) Lama, VN, et al. Prognostic Value of Desaturation during a 6-Minute Walk Test in Idiopathic Interstitial Pneumonia Am. J. Respir. Crit. Care Med., November 1, 2003; 168(9): 1084 - 1090. Abstract
(8) Flaherty, KR, et al. Idiopathic Pulmonary Fibrosis: Prognostic Value of Changes in Physiology and Six-Minute-Walk Test Am. J. Respir. Crit. Care Med., October 1, 2006; 174(7): 803 - 809. Abstract
(9) Lynch DA, et al. Idiopathic Interstitial Pneumonias: CT Features. Radiology; 2005; 236: 10-21. Free Full Text
(10) Johkoh T, et al.IdiopathicIntersitial Pneumonias: Diagnostic Accuraxy of Thin-Section CT in 129 Patients.Radiology 1999; 211: 555-560. Abstract Free Full Text
(11) K.R. Flaherty, G.B. Toews, W.D. Travis, T.V. Colby, E.A. Kazerooni, B.H. Gross, A. Jain, R.L. Strawderman, III, R. Paine, A. Flint, J.P. Lynch, III, and F.J. Martinez Clinical significance of histological classification of idiopathic interstitial pneumonia Eur. Respir. J., Feb 2002; 19: 275 - 283. Free Full Text
(12) Veeraraghavan S, et al. BALfindings in idiopathic nonspecific interstitial pneumonia and usual interstitialpenumonia. Eur Respir J 2003; 22: 239-244. Abstract Free Full Text
(13) Rudd, RM, et al. for the Fibrosing Alveolitis Subcommittee of the British Thoracic Society Study on cryptogenic fibrosing alveolitis: response to treatment and survival Thorax, January 1, 2007; 62(1): 62 - 66. Abstract Free Full Text
(14) Pacanowski MA and Amsden GW. Interferon Gamma-1b in the Treatment of Idiopathic Pulmonary Fibrosis Ann. Pharmacother., October 1, 2005; 39(10): 1678 - 1686. Abstract
(15) Coultas DB, et al. The epidemiology of interstitial lung diseases. Am. J. Respir. Crit. Care Med. 1994; 150: 967-972.
Dr. Holger Klee, 11 / 2007
zum Inhaltsverzeichnis zur Start-Seite
Non-Specific Interstitial Pneumonia NSIP
zum Inhaltsverzeichnis zur Start-Seite
NSIP is one of the more common interstitiall lung diseases. It is to be seen as a heterogene group of diseases, which can partely be secondary to rheumatological or autoimmune diseases. So NSIP has allways to be a priliminary diagnosis and warrants further diagnostic workup.
Patients suffer from dyspnoea, unproductive cough weight loss and some times fever. Lung function reveals diminished diffusion capacity and restriction. The principal findings in high-resolution computer tomography are dissiminated ground glass opacities. Subpleural unaffected humps may be present. In a subgroup of NSIP fibrosing pattern predominate. But the main aspect is again characterized as timely and regionally uniformly affected. The pathophysiological explanation is a proposed „one hit injury“, i.e. the whole lung is injured by a toxin, or anything at one time in all areas. So the radiological and histological findings are equally distributed.
On cytological (broncho-alveolar lavage) or histological examination one can find a lymphocytic infiltration. NSIP is responsive to systemic corticoid steroids. The prognosis is favorable (5 – 10 years).
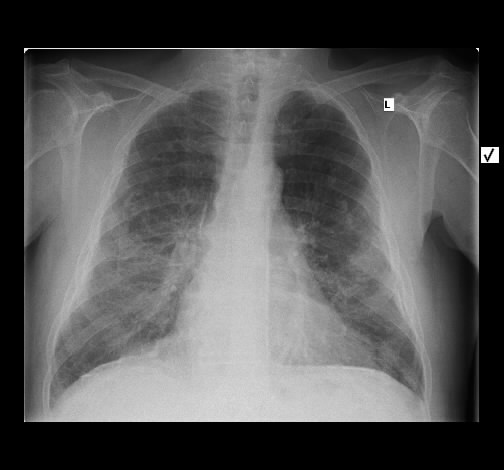
Eine 59-jährige Patientin wurde wegen Reizhusten und Belastungsluftnot in der pneumologischen Ambulanz vorgestellt vorgestellt. Auswurf, Fieber oder Allgemeinverschlechterung bestanden nicht. Die weitere Anamnese war nicht richtungweisend, insbesondere keine Inhalativen Noxen oder Allergene.
Radiologisch stellten sich eine bds. symmetrische flächige Milchgals-Infiltrate dar, zum teil auch fleckig mit subpleuralen Aussparungen. Kein Fibrose, kein Honeycombing.
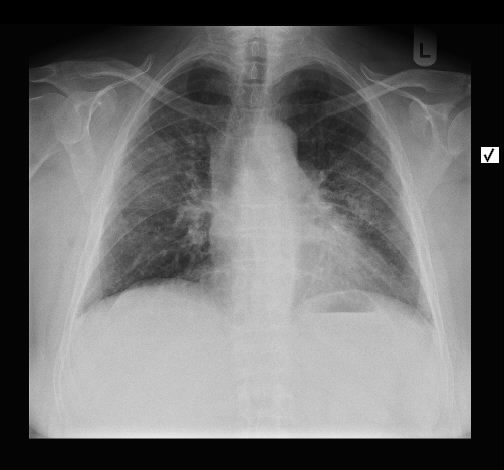
Rö-Thorax: bds. symmetrisch flächige Infiltrate, UL-betont Thorax-CT: symmetrische Michglas-Verschattungen
mit subpleuralen Aussparungen
Das radiologisch Bild ist gut mit einer NSIP vereinbar.
In der Lungenfunktion stellt sich eine mittelgradige Restriktion und diskrete Diffusionsstörung dar.
Lungenfunktion
VC
1.5 l
68 % des Solls
FEV-1
1.3 l
87 % der VC
TLC
2.9 l
71 % des Solls
R(tot)
0.25
TLCO/VA
76 % des Solls
pO2
76 mm Hg
pCO2
40 mmHg
Spiroergometrisch war die Leistungsfähigkeit gemessen an der Sauerstoffaufnahme normal. Eine erhöhte AaDO2 und eine leichte Belastungshypoxämie weist auf eine Diffusionsstörung hin. Weiterhin findet sich eine restriktive Ventilationsstörung.
Spiroergometrie
VO2max.
1213 ml/min
94 % des Solls
VO2/kg
13.8 ml/min/kg
AaDO2
43 mm Hg
pO2min.
59 mmHg
Hinweise auf eine sekundäre interstitielle Lungenerkrankung fanden sich nicht. Auf Vaskulitiden hinweisende Antikörper (ANA, ANCA) waren negativ, anamnestische Hinweise auf eine inhalative Noxe oder ein relevantes inhalatives Allergen bestanden nicht. Präzipitine waren negativ. Eine periphere PE war nicht richtungsweisend. Die BAL ergab eine Lymphozytose. Somit wurde die Diagnose einer NSIP als hochgradig warhscheinlich angesehen und auf eine größere PE mittels VATS verzichtet.
Die Verwendung des Begriffs Non-Specific Interstitial Pneumonia NSIP ist nicht unproblematisch. Zum Einen treten NSIP-artige Histologien im Rahmen von Vaskulititden oder Hypersensitivitäts-Pneumonitiden auf. Daher sollte der histologische Befund einer NSIP-artigen Veränderung immer auch eine breitere Differentialdiagnose veranlassen.
Zum Zweiten stellt die (idiopathische) NSIP eine heterogene Krankheitsgruppe dar, die zum Teil durch unterschiedliche histologische Befunde charakterisiert werden kann. Eine endgültige Klärung ist hier noch nicht erfolgt. Der Begriff NSIP wurde daher als „vorläufig“ in die Klassifikation der interstitiellen Lungenerkrankungen aufgenommen (1).
Die NSIP ist die zweithäufigste idiopathische interstitielle Pneumonitis (2). Das mittlere Erkrankungsalter liegt zwischen 40 und 50 Jahren, auch Kinder können betroffen sein. Die Erkrankungshäufigkeit ist von Geschlecht und Raucherstatus unabhängig (1).
Der klinische Verlauf kann variabel sein. Meist ist der Erkrankungsbeginn langsam protrahiert. Im Vordergrung stehen Dyspnoe, Husten und Abgeschlagenheit, oft auch Gewichtsverlust. Fieber und Trommelschlegelfinger können auftreten. Auskultatorisch kommen trockene Rasselgeräusche vor.
Klinik der NSIP (2)
Dyspnoe
80 %
Husten
33 %
Fieber
22 %
Gewichtsverlust > 6 kg
50 %
Ein langsamer protrahierter Verlauf, ein längerfristig stabiles Krankheitsgeschehen, auch eine vollständige Heilung sind möglich. Die Sterblichkeit ist im Vergleich zur IPF/UIP gering.
Radiologisch imponieren insbesondere bilaterale, symmetrische Milchglas-Infiltrate. Die Verteilung kann auch unregelmäßig sein. Im Gegensatz zu IPF/UIP sind die Befunde jedoch nicht zeitlich heterogen, sondern als typisches Kriterium der NSIP zeitlich uniform. Dem entspricht das histologische Bild. Man spricht vom „one-hit-injury“, was den zeitlich einheitlichen Verlauf der Veränderungen deutlich machen soll.
Radiologische Befunde der NSIP (2)
Milchglas / retikulär
Subpleurale Aussparungen
Traktionsbronchiektasen
Unterlappenschrumpfung
Bilaterale Infiltrate, Unterlappenbetont
Irreguläre Linien
Selten Honeycombing
Sind drei der ersten vier Kriterien gegeben, so wird das radiologische Bild als typisch für eine NSIP angesehen (3).
Histologisch unterscheidet man zwei Ausprägungen. Der zu erwartende klinische Verlauf und das Therapieansprechen ist beim zellulären Typ deutlich günstiger (4,5).
Milde – mäßige interstitielle Entzündung
Homogene interstitielle Fibrose
Typ II Pneumozyten-Hyperplasie
Erhaltene Lungen-Architektur (Elastika-Färbung)
Milde – mäßige interstitielle Entzündung
Keine Fibrosierung
Keine temporale Heterogenität
Keine organisierende Pneumonie
Keine Fibroblasten-Foci
Keine schwere diffuse Entzündung der Alveolar-Septen
Keine acute lung injury (hyaline Membranen)
Wenig Eosinophile
Wenig Granulome
Keine Virus-Einschlüsse oder sonstige Keime
Finden sich in Gewebeproben unterschiedlicher Lokalisation neben NSIP-typischen Veränderungen auch UIP-typische histologische Befunde, so wird die Erkrankung als IPF/UIP klassifiziert. Der klinische Verlauf wird dann voraussichtlich wie eine IPF/UIP sein. Daher wird die Therapie entsprechend sein, wie für diese Erkrankung empfohlen.
Grafik - Prognose der NSIP vs UIP-like NSIP vs UIP (6)
Die broncho-alveoläre Lavage zeigt in der Hälfte der Fälle eine deutliche Lymphozytose, eine Neutrophilie und/oder Eosinophilie (1). Eine neuere Studie fand keinen signifikanten Unterschied in der BAL zwischen NSIP und UIP. Hier waren fast ausschließlich fribrotische NSIP und nur drei zelluläre NSIP eingeschlossen, was die Aussagekraft der Studie einschränkt (7).
Es existieren keine kontrollierten prospektiven Therapiestudien zur NSIP. Empfohlen wird eine Therapie mit systemischen Steroiden. Bei unzureichendem Ansprechen ist eine Immunsuppression mit Azathioprin oder Cyclophosphamid eine Alternative. Es kann mit einem guten Ansprechen und (insbesondere im Vergleich zur UIP) mit einer guten Prognose quo ad vitam gerechnet werden (8). Das 5-Jahres-Überleben liegt um 80% (2).
Initial wurde nach Diagnosestellung eine systemische Steroidtherapie, beginnend mit 50 mg Perdnisolonäquivalent pro Tag begonnen. Hierunter kam es zu einer deutlichen klinischen Besserung, so dass eine Reduktion auf eine Erhaltungsdosis von 5 mg/d möglich wurde. Im weiteren Verlauf ist die Patientin nach 11 Jahren stabil. Der radiologische Befund hat sich kaum verändert. Milchgals-Verschattungen stellen sich nach 10 Jahren diskret rückläufig dar. Es finden sich jetzt diskrete irreguläre Linien und angedeutet verdickte peribronchiale Septen. Lungenfunktionswerte sind auch nach 11 Jahren unverändert. Zusammenfassend handelt es sich um einen typischen über viele Jahre stabilen Verlauf einer NSIP vom zellulären Typ.
(1) American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias . Am. J. Respir. Crit. Care Med. 2001;165: 277-304. Free Full Text PDF
(2) Flaherty KR, et al. Clinical significance of histological classification of idiopathic interstitial pneumonia. Eur. Respir. J., Feb 2002; 19: 275 - 283.
(3) Costabel U, Guzman J. in Kirsten D, Costabel U (Hrsg.) Seltene Lungenkrankheiten 3 – Nicht-spezifische interstitielle Pneumonie interpneu-Verlag, Großhansdorf (2005) S. 133ff.
(4) Katzenstein ALA, Fiorelli RF. Non-specific interstitial pneumonia/fibrosis: histologic patterns and clinical significance. Am. J. Surg. Pathol. 1994; 18:136-267.
(5) Katzenstein ALA, Fiorelli RF. Idiopathic Pulmonary Fibrosis. State of the Art. Am J Respir Cirt Care Med (1997) 157:1301-1315. Free Full Text PDF
(6) Martinez FJ. Idiopathic Interstitial Pneumonias - Usual Interstitial Pneumonia versus Nonspecific Interstitial Pneumonia. The Proceedings of the American Thoracic Society (2006) 3:81-95. Free Full Text
(7) Veeraraghava S, et al. BAL finding in idiopathic nonspecific interstitial pneumonia and usual interstitial pneumonia. Eur Respir J (2003); 22: 239-244. Abstract Free Full Text PDF
(8) Flaherty KR, et al. Radiological versus histological diagnosis in UIP and NSIP: survival implications. Thorax, Feb 2003; 58: 143 - 148. Abstract
Dr. Holger Klee, 9 / 2007
zum Inhaltsverzeichnis zur Start-Seite
Respiratory Bronchiolotis Interstitial Lung Disease RB-ILD
zum Inhaltsverzeichnis zur Start-Seite
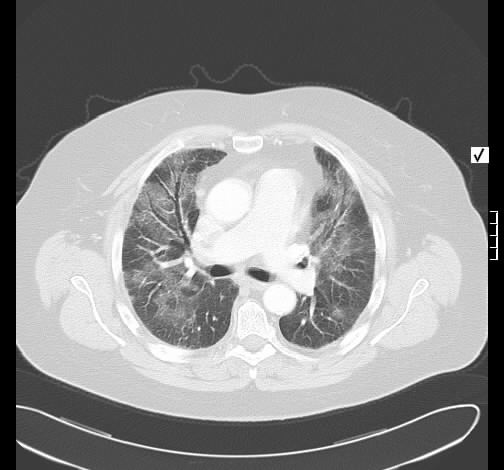
Ein 71-jähriger Raucher (45 pack-years) wird wegen zunehmender Dyspnoe stationär eingewiesen. Es wird insbesondere morgendlicher Husten, jetzt vermehrt mit Expektoration von glasigem Sputum beklagt. Weiterhin ist eine hypertensive herzerkrankung mit Vorhofflimmern und Z.n. embolischem Media-Insult bekannt. Klinisch besteht bds. ein hypersonorer Klopfschall und mittelgradiges Giemen, keine Rasselgeräusche, diskrete Lippenzyanose, keine Temperaturerhöhung.
Radiologisch stellt sich eine beidseite, unterlappenbetonte vorwiegend streifige bis fleckige Zeichnungsvermehrung dar. Im HR-CT finden sich eine felckige, zentrilobuläre Milchgals-Verschattungen, die gut mit einer RB-ILD vereinbar sind.
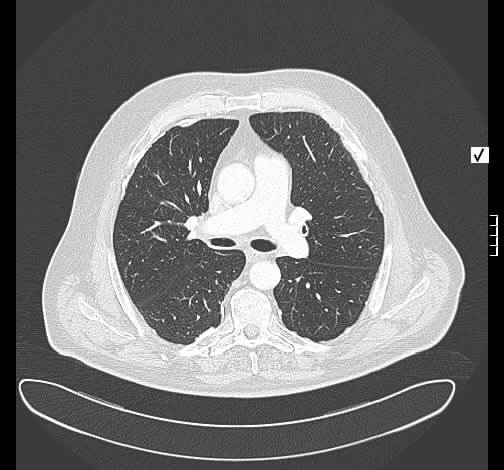
Rö-Thorax: streifige bis fleckige Zeichnungsvermehrung UL Thorax-CT: fleckige, zentrilobuläre Milchglas-Verschattungen
Die Lungenfunktion zeigt eine mittelgradige Obstruktion und leichte Überblähung. Die Diffusion sind leichtgradig beeinträchtigt. Es besteht eine mäßige respiratorische Partialinsuffizienz.
Lungenfunktion
VC
3.2 l
78 % des Solls
FEV-1
1.9 l
58 % des Solls
ITGV
4.2 l
124 % des Solls
R(tot)
0.61
TLCO/VA
77 % des Solls
pO2
59 mm Hg
pCO2
33 mmHg
Die RB-ILD kommt nur bei Rauchern vor. Das Erkrankungsalter liegt meist um 40 Jahre und bei einer Nikotinanamnese von mehr als 30 pack-years. Sind jüngere Raucher betroffen, so liegt der tägliche Nikotinkonsum bei mehr als zwei Schachteln pro Tag (1). Die Häufigkeit der RB-ILD ist proportional zur Raucher-Anamnese (2). Männer sind häufig betroffen (2:1), was wahrscheinlich die Rauchgewohnheiten widerspiegelt (1).
Grafik – pack-years – Häufigkeit der RB-ILD (2)
Es bestehen Beziehung zur Desquamativen Interstitiellen Pneumonitis DIP (1). Aber auch zur pulmonalen Langerhans-Granulomatose bestehen Überlappungen (2).
Grafik – Überlappungen RB – RB-ILD-PLCH-DIP (2)
Es besteht in der Regel eine milde und langsam progrediente Klinik mit Belastungsdyspnoe und unproduktivem Husten. Die Diffusion kann eingeschränkt sein. Eine leichte Obstruktion und Restrikion ist möglich. Oft besteht bei den Rauchern gleichzeitig ein zentrilobuläres Emphysem mit Überblähung (1).
Führender radiologischer Befund der RB-ILD ist eine Verdickung der zentralen Bronchialwände und Milchglas-Verschattungen. Im HR-CT imponieren zentrilobäre Knötchen und fleckige Milchglas-Verschattungen. Ein Emphysem kann gleichzeitig vorliegen (1).
Typisches HR-CT mit zentrilobulären Milchglas-Noduli (6) HR-CT mit fokalem Milchglas und zentrilobulären Noduli (6)
Radiologische Befunde der RB-ILD (1)
Bronchialwand-Verdickung
Fleckig verteiltes Milchglas
Zentrilobuläre Knötchen
Evtl. zentrilobuläres Emphysem in den Oberlappen
HR-CT-Differentialdiagose der Rauchen-assoziierten ILD
Histologisch stellt sich insbesondere eine fleckige und bronchiolozentrische Akkumultion von Alveolarmakrophagen dar. Eine fortgeschrittene interstitielle Fibrose mit Homeycombing patß nicht zum typischen Bild der RB-ILD (1).
Histologie mit pigmentierten Makrophagen und verbreiterter Wand des Bronchus resp. (6)
Fleckige und bronchiolozentrische Verteilung
Bronchozentrische Akkumulation von Alveolarmakrophagen
Milde bronchioläre Fibrose und chronische Entzündung
Makrophagen mit feinen braunen Granula im Zytoplasma
Keine diffuse Makrophagen-Akkumulation
Keine interstitielle Fibrose / kein Honeycombing
Die broncho-alveoläre Lavage ist gelb-braun verfärbt, was auf die Pigmentbeladung der Alveolarmakrophagen hinweist. Bei deutlich erhöhter Gesamtzellzahl ist deren Verteilung normal (3).
Wird das Rauchen eingstellt, so können radiologische Veränderungen und klinische Beschwerden rückläufig sein (5). Jedoch nur 28% der Pateinten erfährt eine klinische Besserung. (4) Die Prognose ist gut. Die 7-Jahres-Überlebensrate liegt bei 75%. Eine systemische Steroidtherapie ist wirksam, aber in der Regel nicht notwendig.
Wegen gleichzeitig bestehender exazerbierter COPD wurde initial eine kurzfristige systemische Steroidtherapie gegeben. Nach Stabilisierung war eine Entlassung mit kombinierter inhalativer antiobstruktiver Medikation (Formoterol, Thiotorpium) möglich. Der intensive Nikotinkonsum konnte beendet werden. In ambulanten Verlaufskontrollen stellte sich die radiologischen Verschattungen rückläufig dar. Es verblieb eine grenzwertige respiratorische Partialinsuffizienz.
(1) American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias . Am. J. Respir. Crit. Care Med. 165: 277-304. Free Full Text PDF
(2) Vassallo R, Jensen EA, et al. The Overlap Between Respiratory Bronchiolitis and Desquamative Interstitial Pneumonia in Pulmonary Langerhans Cell Histiocytosis: High-Resolution CT, Histologic, and Functional Correlations. Chest, October 1, 2003; 124(4): 1199 - 1205. Abstarct Free Full Text PDF
(3) Goeckenjan. R-BILD. In: Kirsten, Costabel Seltene Lungenkrankheiten 1, Großhansdorf: Inter-Pneu-Verlag, 2002: 75ff.
(4) Portnoy J, Veraldi KL, et al. Respiratory Bronchiolitis-Interstitial Lung Disease: Long-term Outcome. Chest, March 1, 2007; 131(3): 664 - 671. Abstract
(5) M. Nakanishi M, Demura Y, et al. Changes in HRCT findings in patients with respiratory bronchiolitis-associated interstitial lung disease after smoking cessation. Eur. Respir. J., March 1, 2007; 29(3): 453 - 461. Abstract
(6) Lynch DA, et al. Idiopathic Interstitial Pneumonias: CT Features. Radiology 2005; 236: 10-21. Lynch DA, et al. Idiopathic Interstitial Pneumonias: CT Features. Radiology 2005; 236: 10-21. Free Full Text
Dr. Holger Klee 9 / 2007
zum Inhaltsverzeichnis zur Start-Seite
zum Inhaltsverzeichnis zur Start-Seite
Ein 33-jähriger Mann stellt sich vor wegen unspezifischem thorakalem Druckgefühl. Ansonsten bestanden keine Beschwerden. Laborparameter waren sämtliche im Normbereich. CT-radiologisch stellt sich eine Raumforderung im Mediastinum rechts dar. Die Raumforderung hat ein unregelmäßiges Enhancement nach Kontrastmittelgabe. Die weitere nicht-invasive Diagnostik ergab keinen richtungsweisenden Befund.
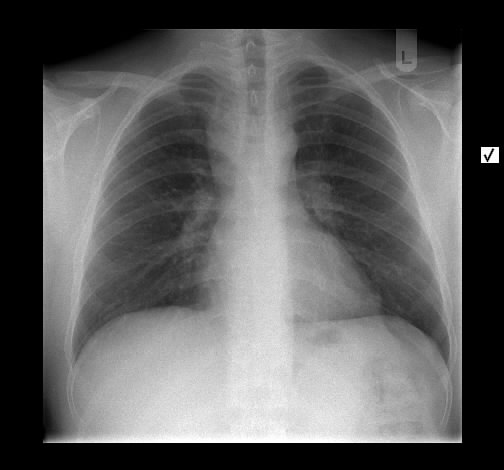
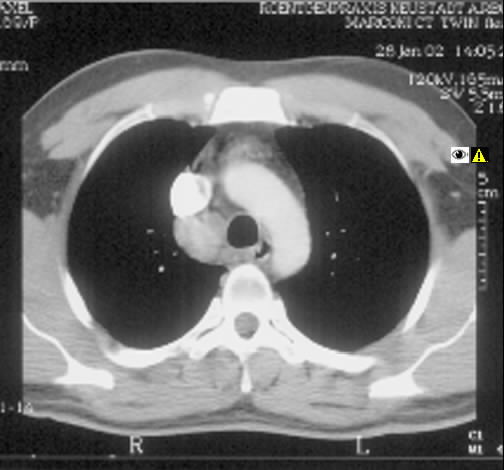
Rö-Thorax: Verbreiterung des Mediastinums re Thorax-CT: LK re mit unregelm. Kontrast
Es erfolgte unter dem Verdacht einen malignen Lymphoms eine Histologiegewinnung per video-assistierter Thorakoskopie. Histologisch stellte sich ein Lymphknoten bestehen aus follikulären und nodulären Proliferaten dar. Um ausgebrannte zellarme Keimzentren liegen konzentrisch, zwiebelschalenartige Schichten von Lymphozyten. In den Keimzentren PAS-positive Fasern und Gefäßproliferate mit epitheloidem Endothel. Es wurde die Diagnose eines Morbus Castleman vom hyalin-vaskulären Typ gestellt.
Bei dem Morbus Castleman handelt es sich um eine sehr seltene Erkrankung mit bislang nicht geklärter Lymphknotenproliferation (1,2). Man unterscheidet eine lokalisierte von einer multizentrischen Form. Die lokalisierte Form tritt eher bei jüngeren Patienten auf, ist oligosymptomatisch und hat eine gute Prognose (1,3).
Lokalisierter vs. multizentrischer M. Castleman (1,3)
12 – 72
19 – 85
Altersmedian
23.5
56
Symptome
evtl. Verdrängung
Fieber, Gewichtsverlust,Juckreiz
Lymphknoten
Zentral
Peripher
Nein
Häufig
Verlauf
Gutartig
Aggressiv
Therapie
Operation
Chemotherapie, (Radiatio ?)
Prognose
5-Jahres-Überleben 100 %
Median 26 Monate
Eine Reihe anderer Organbeteiligungen, wie chylöser Pleuraerguß (4,5) oder BOOP (6) sind beschrieben, jedoch sehr selten.
Die pathogenetische Zuordnung des M. Castleman ist bislang noch nicht sicher. Eine chronisch entzündliche Reaktion wird diskutiert. Hierbei wird eine Assoziation zur HHV-8-Infektion und zum Kaposi-Sarkom beobachtet. Insbesondere die multizentrische Form des M. Castleman kommt gehäuft bei HIV-positiven Patienten vor. Übergänge von multizentrischen Verlaufsformen zu malignen Lymphomen werden beobachtet. (7)
Histologisch unterscheidet man einen hyalin-vaskulären von einem Plasmazell-Typ und eine weitere Mischform (8). Der hyalin-vaskuläre Typ überwiegt bei weitem mit 80 bis 90 % der Fälle (3,5,8). Der hyalin-vaskuläre Typ tritt überwiegend thorakal auf, die Plasmazell-Variante eher intraabdominell. Die Mischform findet sich häufig in peripheren Lymphknotenstationen und zeichnet sich durch einen aggresiven Verlauf mit schlechter Prognose trotz Chemotherapie aus (3).
Histologie hyalin-vaskulärer Typ Histologie Plasmazell-Typ
Histologische Typen des M. Castleman (3)
Plasmazell
Gemischt
33
22
60
Lokalisation
Mediastinum
Abdomen
Periphere LK
Symptome
< 10 %
80 – 90 %
Immer
Operation
Operation
Chemotherapie
Prognose
5-Jahre 100 %
Gutartig
Letalität 50 – 60 %
Die Diagnostik kann erhebliche Schwierigkeiten bereiten (3). Differentialdiagnostisch kommen neben malignen Lymphomen auch nicht-maligne Lymphknoten-Vergrößerungen in Betracht, insbesondere bei HIV-Patienten. Eine definitive Diagnosestellung ist in aller Regel nur durch größere Gewebebiopsien möglich.
Konventionell-radiologisch imponiert insbesondere der lokalisierte mediastinale Typ als hiläre oder mediastinale Lymphadenopathie und damit uncharakteristisch (9,10).
Im Computertomogramm zeigt sich eine deutliche Hypervaskularisation, die sich in der Angiographie wiederfindet mit homogener Kapillarphase (9).
Auch im MRT läßt sich die Hypervaskularisation gut darsetllen, insbesondere beim hyalin-vaskulären Typ. Eventuell ist das MRT zur Abgrenzung benachbarter Strukturen vozuziehen (10). Die Überlegenheit der MRT steht jedoch zur Diskussion (3).
Die beschriebene gute Gefäßversorgung wird zum Teil zum Anlaß genommen, präoperativ eine interventionelle Embolisation vorzunehmen (9).
Bei lokalisiertem Typ führt eine komplette Resektion in der Regel zu einer langfristig rezidivfreien Heilung. Multizentrische Erkrankungen haben eine schlechte Prognose und werden ähnlich wie maligne Lymphome mit Chemotherapie behandelt, z.B. nach dem Schema CHOP. Die Letalität beträgt dennoch 50 – 60 % (3,5).
Der oben angeführte Patient zeigt eine spontane Regression der möglicherweise verbliebene Reste der Lymphadenopathie. Fünf Jahre nach Operation ist der Patient beschwerdefrei. Radiologisch ist bislang kein Rezidiv dokumentiert.
(1) Castleman B, Iverson L, Menendez V. Localized mediastinal lymph-node hyperplasia resembling thymoma. Cancer 1956;9:822-30. pdf
(2) Pontenagel U, Ulrich B. Diagnostik und Therapie des Morbus Castleman. DMW (2004); 129:953-956. pdf
(3) Shahidi H, Myers JL, Kvale OPA. Castleman’s disease. Mayo Clin Proc 1995; 70:969-77. pdf
(4) Hoor TT, Heawan-Lowe K, et al. A Transitional Variant of Castleman’s Disease Presenting as a ChylousPleural Effusion. Chest (1999); 115:285-288. pdf
(5) Blankenship M, Rowlet J, et al. Giant Lymph Node Hyperplasie (Castleman’s Disease) Presenting With Chylous Pleural Effusion. Chest (1997); 112:1132-33. pdf
(6) Wiebel M, Schulz MR, Schulz V. Bronchiolitis obliterans bei paraneoplastischem Pemphigus infolge Morbus Castleman, zwei Fallberichte. Pneumologie 2004; 58. Full Text
(7) Marietta M. Acquired haemophilia in HIV negative, HHV8 positive Multicentric Castleman’s Disease a case report. Abstract
(8) Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other localisations. Cancer 1972;29:670-83.
(9) Meisel S, Rozenman J, et al. Castleman’s Disease – An Uncommon Computed Tomographic Feature. Chest (1988); 93:1306-1307. Free Full Text
(10) Khan J, von Sinner W, et al. Castleman’s Disease of the Chest – Magnetic Resonance Imaging Features. Chest (1994); 105:1608-10. pdf
(11) Castlemans Disease Homepage
Dr. Holger Klee, 8 / 2007
zum Inhaltsverzeichnis zur Start-Seite
zum Inhaltsverzeichnis zur Start-Seite
Ein 69-jähriger Patient beklagte eine allgemeine Schwäche, Gewichtsabnahme von ca. 20 kg, subfebrile Temperaturen und eine längerfristige Heiserkeit. In der Vorgeschichte war eine koronare Herzerkrankung mit Z.n. ACVB-Operation bekannt. Wegen intermittierender Tachyarrhythmie mit Z.n. thrombembolischem cerebralem Insult wurde eine antiarrythmische Therapie mit Amiodaron durchgeführt.
Bei der klinische Untersuchung fielen tastbar vergrößerte Lymphknoten submandibulär auf. Radiologisch imponierten bds. Pleuraergüsse und eine Verbreiterung des oberen Mediastinums, keine pulmonalen Infiltrate.
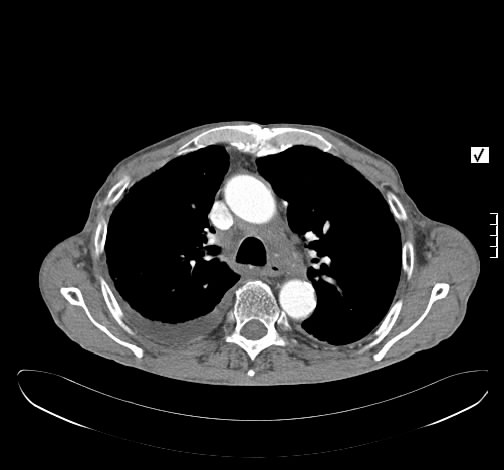
Rö-Thorax: Pleuraergüsse, breites Mediastinum Thorax-CT: LK-Vergrößerung mit inhomog. Kontrastierung, Pleruaergüsse
Im Labor stellte sich eine Hypergammaglobulinämie dar, sonst war das Labor unauffällig. Eine Echokardiographie war abgehen von einer diastolischen Funktionsstörung normal.
Lungenfunktionell stellte sich eine leichte Restriktion, eine leichte Obstruktion und eine allenfalls leichte Diffusionsstörung dar.
Lungenfunktion
VC
2.9 l
60 % des Solls
FEV-1
1.6 l
55 % des Solls
ITGV
4.6 l
118 % des Solls
R(tot)
0.2
TLCO/VA
72 % des Solls
pO2
71 mmHg
pCO2
42 mmHg
Die Punktion der Pleuraergüsse ergab ein Transsudat ohne weiteren richtungsweisenden Befund. Eine Bronchoskopie mit transbronchialer Nadelaspirations-Zytologie eines mediastinalen Lymphknotens ergab keinen pathologischen Befund.
Differentialdiagnostisch kam auch eine seltene pulmonale Nebenwirkung von Amiodaron in Frage. Zur weiteren Klärung wurde eine Mediastinoskopie durchgeführt. Hierbei war die Blutstillung ungewöhnlich aufwendig. Bei M. Castleman wird zum Teil vor resektiven Maßnahnmen eine Katheteremobilsation durchgeführt.
Histologisch stellten sich in den mediastinalen Lymphknoten mittelgroße Keimzentren dar, welche von zwiebelschalenartigen Schichten aus kleinen ausgereiften Lymphozyten umgeben werden. In den Keimzentren sowie im parakortikalen Bereich dünnwandige PAS-positive Kapillaren. Weiterhin kommen hyperplastische Blutgefäße zur Darstellung. Der Befund ist gut mit einem hyalin-vaskulären Typ des M. Castleman vereinbar.
Histologie hyalin-vaskulärer Typ
Zur ausführlichen Beschreibung des M. Castleman verweise ich auch den obenstehenden Case Report eines M. Castleman vom lokalisierten Typ.
Thorakale Manifestationen
Neben einer mediastinalen Lymphknotenvergrößerung sind weitere thorakale Krankheitsmanifestationen möglich. Das Auftreten eines Pleuraergusses (1,2) bzw. einer BOOP-Reaktion (3) bei M. Castleman ist bei multilokulärem Typ nicht selten. Ein Pleuraerguss wurde bei 3 von 12 Patienten beschrieben (4). Eine retikuläre bzw. noduläre Zeichnungsvermehrung fand sich bei je 7 von 12 (4), im HR-CT bei 12 von 12 Patienten (5). Ein Pleuraerguß kann als Chylothorax auftreten (1,2) .
Bei unserem Patienten handelte es sich bei den bds. Pleuraergüssen um ein reines Transsudat. Das Ansprechen auf eine hochdosierte Steroidtherapie spricht dafür, dass diese Krankheitsmanifestationen im Rahmen des M. Castleman zu sehen sind. Auch eine rein pleurale Manifestation des M. Castleman ist beschrieben (6). Bei dem HHV-8-assoziierten pleural effusion lymphoma besteht im Gegensatz zum M. Castleman und zum Kaposi-Sarkom eine Co-Infektion mit EBV und eine Expression von CD30 (7).
Im Verlauf stellte sich vor endgültiger histologischer Diagnosesicherung eine pulmonale Zeichungsvermehrung dar, so dass unter dem Verdacht einer BOOP-Reaktion bereits eine Steroid-Therapie eingeleitet wurde. Hiervon profitierte der Patient subjektiv. Infiltrate und Pleruaergüsse waren rückläufig.
Die therapeutischen Optionen bei multilokulären M. Castleman wird als Erstlinientherapie Interferon-Alpha empfohlen (8,9). Die Therapie umfassen weiterhin Chemotherapie, wobei Etoposid / Doxorubicin wirksamer als CHOP ist, hochdosierte Steroide, Immunsuppressiva und Anti-CD20-Faktoren (Rituximab) (10,11,12) und IL-6-Antagonisten (13). Insbsondere bei HIV-Patienten ist M. Castleman regelhaft mit HHV8 / KSHV assoziiert, so dass eine Therapie mit Ganciclovir in Frage kommt (14). Bei multilokulärem Befall ist eine Operation mit kurativem Ansatz nicht möglich.
Wegen des multilokulären und symptomatischen Krankheitsausprägung ist von einer eingeschränkten Prognose auszugehen. Es wurde eine Chemotherapie eingeleitet.
(1) Hoor TT, Heawan-Lowe K, et al. A Transitional Variant of Castleman’s Disease Presenting as a Chylous Pleural Effusion. Chest (1999); 115:285-288.
(2) Blankenship M, Rowlet J, et al. Giant Lymph Node Hyperplasie (Castleman’s Disease) Presenting With Chylous Pleural Effusion. Chest (1997); 112:1132-33. Free Full Text
(3) Wiebel M, Schulz MR, Schulz V. Bronchiolitis obliterans bei paraneoplastischem Pemphigus infolge Morbus Castleman, zwei Fallberichte. Pneumologie 2004; 58. pdf
(4) Guitot A, et al. Pulmonary manifestations of multicentric Castlemans disease in HIV infection: a clinical, biological and radiological study. Eur. Respr. J., 2005; 26(1):118-25. Abstract Free Full Text
(5) Johkoh J, et al. Intathoracic multicentric Caslteman disease: CT findings in 12 patients.Radiology, 1998; 209(2): 477-81. Abstract Free Full Text (pdf)
(6) Ko SF, Ng SH, et al. Castleman disease of the pleura: experience with eight surgically proven cases. Ann. Thorac. Surg., 2003; 76(1): 219-24. Asbtract
(7) Hengge UR, et al. Update on Kaposis sarcoma and other HHV associated diseases. Part 2: pathogenesis, Castlemans disease, and pleural effusion lymphoma. Lancet Infect Dis, 2002; 2(6): 344-52. Abstract Full Text
(8) Andres E, Maloisel F. Interferon-alpha as first-linke therapy for treatment of multicentric Caslemans disease. Ann. Onc. 2000;11(12):1613-4. Abstract
(9) Nord JA, Karter D. Low dose interferon-alpha therpy for HIV-associated multicentric Castlemans disease. Int J STD AIDS, 2003;14(1): 61-2. Abstract
(10) Pontenagel U, Ulrich B. Diagnostik und Therapie des Morbus Castleman. DMW (2004); 129:953-956. pdf
(11) Laurence G, et al. Prospective Study of Rituximab in Chemotherapy-Dependent Human Immunodeficiency Virus-Associated Multicentric Castlemans Disease: ANRS 117. JCO 2007: 3350-3356. Abstract
(12) Gerard L, et al. Prospective study of rituximab in chemotherpy-dependent human immunodeficiency virus associated multicentric Castlemans disease: ANRS 117 CastlemaB Trial. Abstract
(13) Nishimoto N, et al. Improvement in Caslemans disease by humanized anit-interleukin-6 receptor antibody therapy. Blood 2000; 95(1): 56-61. Abstract Free Full Text
(14) Csper C, et al. Remission von HHV-8 and HIV-associated multicentric Castlman disease with ganciclovir treatment. Blood, 2004; 103(5):1632-4. Abstract Free Full Text
(15) Castlemans Disease Homepage
Dr. Holger Klee, 8 / 2007
zum Inhaltsverzeichnis zur Start-Seite
zum Inhaltsverzeichnis zur Start-Seite
Eine 49-jährige Patientin stellte sich vor wegen zunehmender Belastungsdyspnoe und geringgradigem Reizhsuten. Keine Schmerzen, keine Allgemeinverschlechterung, kein Fieber oder Auswurf. Bei der körperlichen Untersuchung fand sich bds. basal eine Klopfschalldämpfung von ca. 5 Querfinger und dort ein abgeschwächtes Atemgeräusch. Die Fingernägel waren verdickt und gelblich. Der übrige körperliche Untersuchungsbefund war unauffällig.
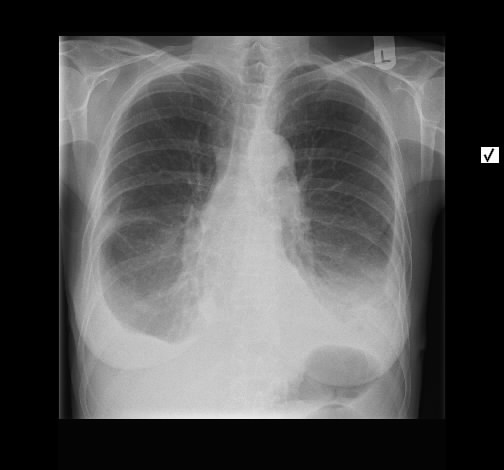
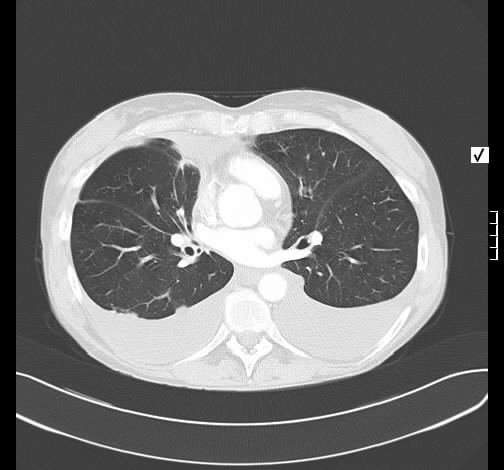
Rö-Thorax Thorax-CT
Bild 1 Bild 2 Bild 3 Bild 4 Bild 5
gelb-verfärbte und verdickte Nägel
Radiologisch stellten sich beidseitige Pleuraergüsse dar. Das Pleurapunktat ergab ein Exsudat mit normalem Zuckergehalt. Die zytologische Untersuchung ergab kein Hinweis auf Malignität. Bakteriologische und mykobakteriologische Kulturen blieben steril. Ein Tuberkulin-Haut-Test mit 10 TE war negativ.
Somit wurde die Diagnose eines Yellow-Nail-Syndromes gestellt.
Das Yellow-Nail-Syndrom wird klassischerweise durch die Trias gelb-verfärbter, verdickter Nägel, Pleuraergüsse und Lymphödeme charakterisiert (1,2,3,4,5). Weiterhin können Bronchiektasen und rezidivierende Sinusitiden auftreten. Das Auftreten der einzelnen Symptome muß jedoch nicht zeitgleich geschehen (6).
Bei dem Yellow-Nail-Syndrom handelt es sich um eine seltene Erkrankung (ca. 1: 500.000 – 1: 1.000.000) - wobei Frauen etwas häufiger betroffen sind - mit bislang nicht eindeutig geklärter Ätiologie. Das Syndrom kann als Paraneoplasie auftreten bei unterschiedlichsten malignen Erkrankungen (7,8,9). Aber auch in Assoziation zu Immunerkrankungen (3,10), rheumatologischen, endokrinologischen , Nephritiden und Tuberkulose (11) wurde das Syndrom beschrieben. Am häufigsten handelt es sich jedoch um ein idiopathisches Krankheitsbild mit guter Prognose ohne nachweisbare Grunderkrankung. Eine differentialdiagnostische Klärung ist daher zu empfehlen.
Pathophysiologisch wird eine gestörte Lymphdrainage diskutiert (12). Möglicherweise ist auch ein Protein-Verlust an der Ausbildung der Pleuraergüsse beteiligt (13).
Die gelblicher Verfärbung und Verdickung der Fingernägel ist Folge eines verlangsamten Nagelwachstums (14).
Therapeutisch sind meist entlastende Pleurapunktionen nötig. Bei häufig notwendigen Entlastungspunktionen ist eine Pleurodese mit Talkum effektiv (15). Als seltene Therapiealternative kommt auch ein pleuro-peritonealer Shunt in Frage (16). Auch eine physiotherapeutische Lymphdrainage und Diätmaßnahmen wurden als effektiv beschrieben. Bericht einer Patientin
Die Nagelveränderungen können bei effektiver Therapie einer Grunderkrankung gut rückläufig sein (7,8). An Ansprechen auf eine lokale oder systemische Therapie mit Vitamin E wurde beschrieben (6).
Bei der oben beschriebenen Patientin wurde wiederholt entlastende Pleurapunktionen notwendig. Daher wurden sequentiell beidseits Pleurodesen mit Talkum durchgeführt. Nach 7-jährigem stabilem Krankheitsverlauf mußte die Pleurodese wiederholt werden. Der Allgemeinzustand der Patientin ist weiterhin gut.
Literatur
(1) Emerson, PA (1966) Yellow nails, lymphedema and pleural effusion. Thorax 21,247-253
(2) Samman, PD, White, WF (1964) The ’yellow nail syndrome.’ Br J Dermatol 76,153-157
(3) Siegelman SS Lymphedema, Pleural Effusion and Yellow Nails: Associated Immunologic Deficiency Chest 1969;56: 114-117. Free Full Text
(4) Riedel, M. Multiple Effusions and Lymphedema in the Yellow Nail Syndrome. Circulation. 2002;105:e25. Free Full Text
(5) Alkadhi, H, et al. Yellow Nail Syndrome. Respiration 2005;72:197 Free Full Text (PDF)
(6) M. Riedel M, Bohanes, V. Das Yellow-Nail-Syndrom. Pneumologie 2003; 57: 144-148 Full Text
(7) P. Steve, P, et al. Skin Lesions in Malignancy - Case 3. Yellow Nail Syndrome in Non-Hodgkins Lymphoma. Journal of Clinical Oncology, Vol 19, Issue 7 (April), 2001: 2100-2101. Free Full Text
(8) Mobeen I, et al. Yellow Nail Syndrome-Resolution of Yellow Nails After Successful Treatment of Breast Cancer Chest. 2000;117:1516-1518. Free Full Text
(9) Thomas PS, Sidhu B. Yellow nail syndrome and bronchial carcinoma. Chest, 1987; 92: 191. Free Full Text
(10) Bokszczanin A, Levinson A. Coexistent yellow nail syndrome and selective antibody deficiency. Ann Allergy Asthma Immunol, November 1, 2003; 91(5): 496-500. Abstract
(11) Prasad TR. Yellow nail syndrome Chest, 1980; 77: 580. Free Full Text
(12) RH Bull, DA Fenton, and PS Mortimer. Lymphatic function in the yellow nail syndrome. Br J Dermatol, February 1, 1996; 134(2): 307-12. Abstract
(13) DAlessandro, A, et al. Yellow nail syndrome: does protein leakage play a role? European Respiratory Journal 17:149-152 (2001). Free Full Text
(14) Moffitt, de Bekrer R. Yellow nail syndrome: the nail that grows half as fast grows twice as thick. Clinical and Experimental Dermatology 2000; 25:21. Abstract
(15) Glazer M, Successful Talc Slurry Pleurodesis in Patients With Nonmalignant Pleural Effusion - Report of 16 Cases and Review of the Literature Chest. 2000;117:1404-1409. Free Full Text
(16) Brofman JD, et al. Yellow nails, lymphedema and pleural effusion. Treatment of chronic pleural effusion with pleuroperitoneal shunting Chest 1990; 97: 743-745. Free Full Text (PDF)
Dr. Holger Klee, 5 / 2007
zum Inhaltsverzeichnis zur Start-Seite
Chronische eosinophile Pneumonie CEP
zum Inhaltsverzeichnis zur Start-Seite
Ein 43-jähriger Nichtraucher beklagte eine seit sechs Monaten progrediente Belastungsdyspnoe, sowie wenig glasigem Auswurf und ein allgemeines Krankheitsgefühl. Es bestand keine Temperaturerhöhung, keine Gewichtsabnahme. Bei Aufnahme fand sich bds. in Mittel-Oberfeldern ein feinblasiges Knister, kein Giemen. Der übrige körperliche Status war unauffällig.
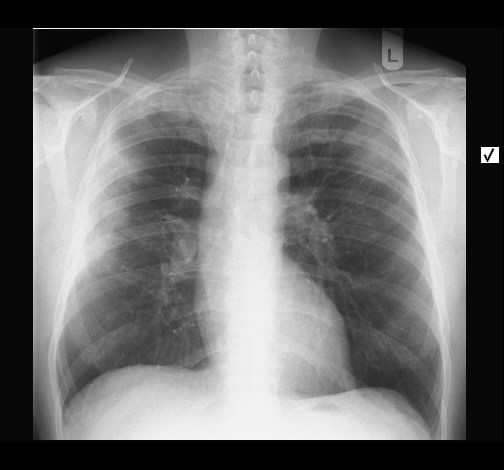
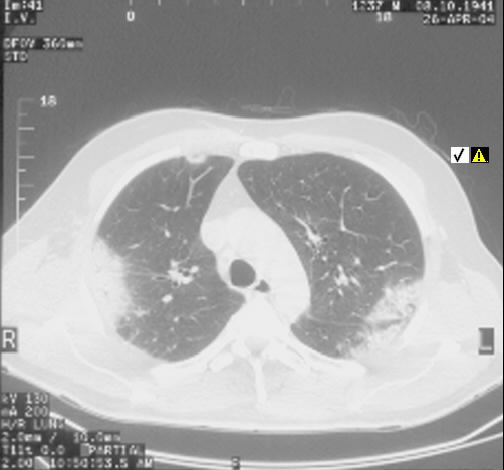
Rö-Thorax: bds. periphere Infiltrate Mittel-Oberfeld Thorax-CT: bds. flaue periphere Infiltrate Mittel-Oberfeld
Radiologisch stellten sich bds. periphere Infiltate dar in den Mittel-Oberfeldern. Im CT kommt die Beidseitigekeit deutlicher zur Darstellung. Die peripher gelegenen Infiltrate erscheinen wolkig, flau begrenzt. Mediastinale Lymphknoten waren nicht vergrößert.
Im Labor bestand eine geringe Leukozytose 10.1 mit deutlicher peripherer Eosinophlie 17 %. C-reaktives Protein war mit 3.6 mg/dl leicht erhöht (Norm bis 1.0). In einer broncho-alveolären Lavage findet sich ebenfalls eine mäßige Eosinophilie von 12 %. Eine transbronchiale periphere Lungen-PE ergab eine eosinophile intraalveoläre Entzündung ohne Hinweis auf Vaskulitis in den angetroffenen Gefäßen.
Es stellt sich die Differentialdiagnose einer eosinophilen Pneumonie. Als Diagnosekriterien werden Infiltrat, pheriphere Eosinophilie und Eosinophilie der BAL erwartet.
Diagnose-Kriterien einer Eosinophilen Pneumonie (1)
radiologisches Infiltrat
periphere Eosinophilie
> 1 x 109 Eos/l
und
Eosinophilie in der BAL
> 25 %
und / oder
Differentialdiagnostisch kommen eine Reihe von Erkrankungen in Frage. Die ältere Einteilung der pulmonalen Eosinophile nach Crofton sollte zugunsten der klinisch praktikablen Klassifikation nach Cottin und Cordier verlassen werden.
Klassifikation der Eosinophilen Lungenkrankheiten (1)
A - Eosinophile Lungenerkrankung unklarer Genese
idiopathisch
Idiopathische chronische eosinophile Pneumonie
Idiopathische akute eosinophile Pneumonie
assoziiert mit Systemerkrankungen
Churg-Strauss-Syndrom
Hypereosinophiles Syndrom
B - Eosinophile Lungenerkrankung bekannter Genese
parasitäre eosinophile Pneumonie
andere infektiöse eosinophile Pneumonie
Allergische bronchopulmonale Mykose
Bronchozentrische Granulomatose
Toxische eosinophile Pneumonie (Medikamente, Toxine, Radiatio)
C - Verschiedene Lungenerkrankungen, evtl. assoziiert mit Eosinophile
Bronchiolitis obliterans mit organisierter Pneumonie
Asthma und eosinophile Bronchitis
Idiopathische interstitielle Pneumonien
Langerhans-Zell-Granulomatose
Lungen-Transplantation
Sarkoidose
Paraneoplastische eosinophile Pneumonie
Hinweise auf eine Systemerkrankung, Infektionserkrankungen, insbesondere Parasitosen, Asthma oder einen anamnestischen Hinweis auf ein Medikatmenten- / Toxin-Expositition lagen nicht vor. Damit konnte die Diagnose einer chronischen eosinophile Pneumonie gestellt werden.
Die chronische eosinophile Pneumonie ist eine seltene Erkrankung des mitteleren Lebensalters (Mittel um 45 Jahre), die vorwiegend Frauen betrifft (F:M = 2:1). Die Erkrankung ist mit Atopie (50 %) und Asthma bronchiale (30 %) assoziiert und anscheinend nur bei Nichtrauchern vorzukommen.
Die Zeit bis zur Diagnosestellung beträgt nach Literaturangaben im Mittel 7.7 Monate.
Neben respirtorischen Symptomen steht bei chronisch eosinophiler Pneumonie oft auch ein allgemeinges Krankheitsgefühl im Vordergrund.
Respiratorische Symptome bei CEP (2)
Husten
90 %
Dyspnoe
60 – 90 %
Auswurf
30 – 40 %
Thoraxschmerz
10 – 15 %
Hämoptysen
< 10 %
Allgemeinsymptome bei CEP (2)
Fieber
70 %
Gewichtsabnahme
50 – 75 %
Müdigkeit
50 %
Nachtschweiß
80 %
Der angeführte Patient berichtete neben einer allgemeinen Abgeschlagenheit keine weiteren Allgemeinsymptome.
Konventionell-radiologisch sind beidseitige periphere Infiltrate typisch, die sich vorwiegend in den Mittel-Oberfeldern finden. Oft wird auch - didaktisch wertvoll - von einem "Negativ-Bild eines Lungenödems" gesprochen. Die Beidseitigekeit läßt sich bisweilen nur im Thorax-CT gut dokumentieren.
Mittels HR-CT läßt sich bei eosinophilen Infiltraten mit recht guter Genauigkeit eine differentialdiagnostische Zuordnung treffen und andere eosinophile Infiltrate abgrenzen. Eine Korrelation zu klinischen Befunden, BAL und Histologie ist jedoch in jedem Fall notwendig.
Typische HRCT-Befunde bei CEP (3,4)
fleckige uni- oder bilaterale Infiltrate
periphere Betonung
Milchglas / Konsolidierung in Mittel-Oberfeldern
bandförmige subpleurale Verdichtungen
Eine Abgrenzung zur BOOP aufgrund radiologischer Kriterien kann schwierig sein. Eine Zusammenschau mit klinischen Befunden, BAL und Histologie ist erforderlich. (2,3)
Differentialdiagnose zwischen CEP und BOOP (5)
flaue Infiltrate peripher
dichtes dreiecksförmiges Infiltrat
Mittel-Oberfeld
Mittel-Unterfeld
selten Aerobronchgramm
häufig Aerobronchogramm
verdickte Septen 72 %
verdickte Septen 40 %
Noduli 5 %
Noduli 32 %
retikuläres Muster 9 %
retikuläres Muster 45 %
Bronchialerweiterung 26 %
Bronchialerweiterung 58 %
peribronchial 9 %
peribronchial 29 %
Eosinophilie > 20 %
buntes Zellbild
keine Fibrose
fibroblastenreich
Schaumzellen selten
Schaumzellen häufig
BO selten
BO häufig
Funktionsdiagnostik
Die Lungenfunktionsdiagnostik ergibt häufig eine restriktive Ventilationsstörung und eine respiratorische Partialinsuffizienz. (2,6) Bei unserem Patienten war die Lungenfunktion vollständig unauffällig.
Lungenfunktion
VC
5.0 l
98 % des Solls
FEV-1
3.9 l
78 % des Solls
ITGV
2.9 l
99 % des Solls
R(tot)
0.11
TLCO/VA
87 % des Solls
pO2
74 mm Hg
pCO2
36 mm Hg
Spiroergometrie
VO2max.
2062
98 % des Solls
VO2/kg
26.1
34
In der Spiroergometrie ließ sich eine Diffusionsstörung und / oder Verteilungsstörung sicher ausschließen.
Es existieren keine kontrollierten oder gar randomisierten Studien zur Therapie der chronischen eosinophilen Pneumonie. Allgemein akzeptierte Empfehlungen geben eine systemische Steroidtherapie vergleichbar mit der Therpie der Sarkoidose an. (2,7) Die initiale Dosis beträgt dabei 0.3 – 0.5 mg Prednisolon/kg täglich. Nach vier Wochen kann eine langsmae Dosisreduktion begonnen werden. Rezidive sind in bis zu der Hälfte der Fälle bei Dosisreduktion zu erwarten. Die Rezidive treten typischerweise mit der selben Lokalisation auf und sprechen gut auf die initiale Therapie an. (2,3) Die Letalität ist sehr gering. Nur Einzelfälle sind bisher berichtet worden. (2)
Der beschriebene Patient erhielt eine systemische Steroidthearpie mit 40 mg pro Tag (0.5 mg/kg) über vier Wochen. Bereits nach wenigen Tagen war die klinische Symptomatik vollständig rückläufig. Die Steroiddosis wurde nach vier Wochen stufenweise reduziert. Ein Rezidiv ist innerhalb von drei Jahren Nachbeobachtungszeit nicht aufgetreten.
(1) Cottin V, Cordier JF Eosinophilic pneumonias Allergy 2005; 60: 841-857. Abstract Free Full Text
(2) Kirsten, D. Idiopathische eosinophile Pneumonien Pneumologie 2002; 56:621-630. Free Full Text
(3) Johkoh T, et al. Eosinophilic Lung Diseases: Diagnostic Accuracy of Thin-Section CT in 111 Patients Radiology 2000; 216: 773. Free Full Text Abstract
(4) Jeong, YJ, et al. Eosinophilic Lung Diseases: A Clinical, Radiologic, and Pathologic Overview RadioGraphics, 2007; 27(3): 617 - 637. Abstract
(5) Arakawa H, et al Bronchiolitis Obliterans with Organizing Pneumonia Versus Chronic Eosinophilic Pneumonia - High-Resolution CT Findings in 81 Patients AJR 2001; 176:1053-1058. Free Full Text Abstract
(6) Fox B and Seed WA Chronic eosinophilic pneumonia Thorax 1980; Vol 35, 570-580. Abstract
(7) Voss, M, et al. Die chronische eosinophile Pneumonie (CEP) Dtsch Med Wochenschr 2004 ;129 :1858-1860. Full Text
Dr. Holger Klee, 6 / 2007
zum Inhaltsverzeichnis zur Start-Seite
Desquamative Interstitielle Pneumonitis DIP
zum Inhaltsverzeichnis zur Start-Seite
Eine 63-jährige Patientin stellte sich wegen zunehmender Belastungsluftnot und unproduktiven Reizhusten vor. Es lag kein Fieber und keine Allgemeinverschlechterung vor. Die Patientin hatte bis vor 3 Jahren geraucht, entsprechend einer Nikotinanamnese von ca. 40 pack years.
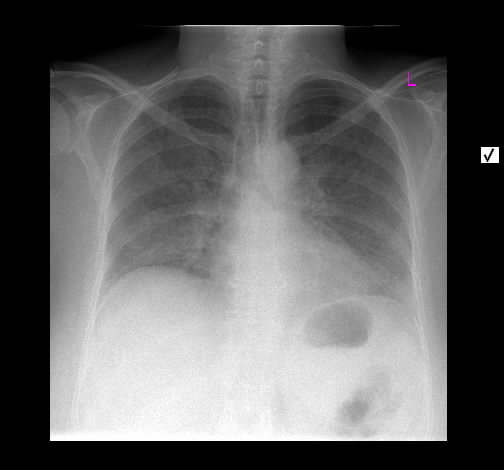
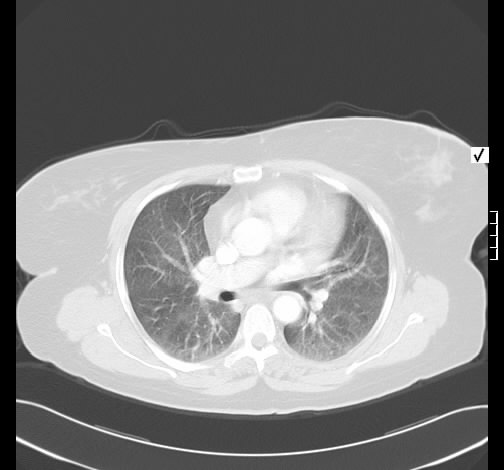
Rö-Thorax: Milchglas-Verschattung bds. basal betont Thorax-CT: bds. ausgedehnte Milchglas-Verschattungen bds. symmetrisch,
basal betont, zum Teil Landkarten-ähnlich
Die weitere Anamnese war nicht richtungsweisend. Keine Haustiere, keine Landwirtschaft, keine außergewöhnliche Schimmelexposition, keine Medikation, keine Begleiterkrankungen.
In der Lungenfunktion fand sich eine schwere Restriktion sowie eine schwere Hypoxämie in Ruhe.
Lungenfunktion
VC
0.9 l
39 % des Solls
FEV-1
0.8 l
88 % der VC
TLC
2.9 l
63 % des Solls
R(tot)
0.31
TLCO/VA
nicht möglich
pO2
45 mm Hg
pCO2
32 mmHg
Die broncho-alveoläre Lavage ergab eine Lymphozytose (15%). Eine periphere Lungen-PE war wegen der initial schweren Hypoxämie und des damit hohen periinterventionellen Risikos nicht möglich.
Differentialdiagnostisch kam zunächst auch eine Exogen Allergische Alveolitis in Frage. Ein auslösendes Agens war nicht zu eruieren. Die Klinik mit eher langsam zunehmender Dyspnoe und fehlender Besserung unter der Allergenkarenz während des stationären Aufenthaltes sprach jedoch gegen diese Differentialdiagnose.
Der klinische Verlauf und das radiologisch Bild sind gut mit einer Desquamativen Interstitiellen Pneumonitis DIP vereinbar.
Die Desquamative Interstitielle Pneumonitis ist eine seltene interstitielle Lungenerkrankung. (29) Genaue epidemiologische Daten zur Inzidenz liegen nicht vor. (1,2) Typisches Manifestationsalter ist die 4. bis 5. Dekade. (3,4) Bei Kindern ist die DIP selten. (5) Ob Männer häufiger betroffen sind, ist strittig. Manche Autoren geben ein Geschlechtsverhältnis M:F von 2:1 an. (3,4)
Die DIP kommt fast ausschließlich bei Rauchern vor. Es besteht eine Ähnlichkeit zu anderen durch Rauchen bedingten Lungenerkrankungen. (1,2,3,4,6,7,8) Nur ca. 10 bis 13 % der Patienten mit DIP haben nie geraucht. (2,3) Weiterhin kann das klinische und radiologische Bild einer DIP bei lymphoproliferativen Erkrankungen (9) und medikamentös-toxisch (10) auftreten.
Meist besteht eine langsam progrediente klinische Symptomatik mit vorherrschender Dyspnoe. Weiterhin können Husten und Thoraxschmerzen auftreten. Knisterrasseln ist häufig. (2,3)
Klinik und Befunde der DIP (2)
Dyspnoe
87 %
Husten
43 %
Thoraxschmerz
17 %
Knisterrassel
57 %
Trommelschlegelfinger
26 %
Lungenfunktionell ist eine Restriktion und Diffusionsstörung typisch, aber keineswegs immer anzutreffen.
Lungenfunktion bei DIP (2)
Restriktion
30 %
Obstruktion
15 %
Diffusionsstörung
35 %
Normalbefund
20 %
Radiologisch sind für eine DIP Milchglastrübungen typisch, die basal und peripher betont sind. Kleine, peribronchioläre, dünnwandige Zysten können vorkommen. (2,11,12)
Typisches HR-CT mit Milchglas und kleinen peribronchovaskulären Zysten (11)
Radiologische Befunde der DIP (2)
Milchglas
100 %
basal betont
73 %
peripher betont
59 %
bilateral symmetrisch
86 %
dünnwandige zysten
32 %
HR-CT-Differentialdiagose der Rauchen-assoziierten ILD (3)
Das HR-CT stellt einen wichtigen Baustein der Diagnostik dar. Eine Diagnosestellung ist in Zusammenschau der Klinik und der radiologischen Befunde und des Verlaufs möglich. (13,14,15,16,17,18)
Eine offene Lungen-PE mittel VATS wird zur Diagnosestellung selten notwendig werden. (2) Das charakteristische Bild der DIP ist eine homogene Verdickung der Alveolarsepten und eine intraalveoläre Akkumulation von Alveolarmakrophagen. (2,3,19,20,23,24,25) – Ursprünglich nahm man fälschlicherweise an es handele sich um eine Desquamation von Alveolarzellen. (21,22) Die irreführende Krankheitsbezeichnung blieb aber bislang erhalten.
Histologie mit Akkumulation von Alveolarmakrophagen und milde interstitielle Verdickung (11)
Histologie der DIP
Intraalveoläre Alveolarmakrophagen
verdickte Alveolarsepten
nur geringe interstitielle Entzündung
keine Fibrosierung
Da eine DIP meist durch Rauchen verursacht wird, sollte Nikotinkarenz eingehalten werden. Unter einer systemischen Steroidtherapie ist in der Regel ein Ansprechen zu erwarten. Rezidive können auf eine erneute Steroidtherapie ansprechen. (26,27) Die Indikation zur immunsuppressiven Therapie, z. B. mit Azathioprin, wir unterschiedlich beurteilt. (3)
Bei ca. 30 % der Patienten muß mit einer mittelfristigen Verschlechterung gerechnet werden. Die 5-Jahres-Letalität liegt bei 25 %. (2)
Möglicherweise kann eine DIP nach Lungentransplanation wiederauftreten.
Es wurde nach Stellen der Verdachtsdiagnose einer DIP eine systemische Steroidtherapie begonnen. Hierunter konnte eine Besserung der klinsichen Beschwerden und der funktionellen Einschränkung erreicht werden. Eine Sauerstofflangzeittherapie wurde eingeleitet.
Im weiteren Verlauf kam es wiederholt zu Rezidiven mit erneut schwerer funktioneller Einschränkung. Es konnte jeweils mit systemischer Steroidtherapie ein erneutes Ansprechen induziert werden. Nach fünfjährigem Verlauf und dem sibeten Rezidiv wurde die immunsuppressive Therapie um Azathioprin ergänzt.
Die Lungenfunktion und das radiologische Bild waren auch nach langjährigem Verlauf noch ohne wesentliche progrediente Verschlechterung.
(1) Demedts M, et al. Interstitial lung diseases: an epidemiological overview. Eur. Respir. J., 2001; 18: 2S - 16s. Free Full Text
(2) Ryu, JH, et al. Desquamative Interstitial Pneumonia and Respiratory Bronchiolitis-Associated Interstitial Lung Disease. Chest, 2005; 127(1): 178 - 184. Abstract
(3) Ryu JH, et al. Smoking-related interstitial lung diseases: a concise review. Eur. Respir. J., Jan 2001; 17: 122 - 132. Free Full Text
(4) Demedts M and Costabel U, ATS/ERS international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Eur. Respir. J., 2002; 19: 794 - 796. Free Full Text
(5) Stillwell PC, et al. Desquamative interstitial pneumonitis in children. Chest, 1980; 77: 165 - 171. PDF
(6) Ryu JH, et al. Desquamative Interstitial Pneumonia and Respiratory Bronchiolitis-Associated Interstitial Lung Disease. Chest, 2005; 127: 178 - 184. Free Full Text
(7) Vassallo R, et al. The Overlap Between Respiratory Bronchiolitis and Desquamative Interstitial Pneumonia in Pulmonary Langerhans Cell Histiocytosis: High-Resolution CT, Histologic, and Functional Correlations. Chest, 2003; 124: 1199 - 1205. Free Full Text
(8) Gould VE, et al. Desquamative Interstitial Pneumonia. Chest, 1971; 59: 349 - 352. PDF
(9) Goldstein JD , Godleski JJ, and Herman PD, Desquamative interstitial pneumonitis associated with monomyelocytic leukemia. Chest, 1982; 81: 321 - 325. PDF
(10) Bone RC, et al. Desquamative interstitial pneumonia following chronic nitrofurantoin therapy. Chest, 1976; 69: 296 - 297. PDF
(11) Lynch DA, et al. Idiopathic Interstitial Pneumonias: CT Features. Radiology 2005; 236: 10-21. Free Full Text
(12) Hartman TE, et al. Desquamative interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1993; 187: 787. Abstract PDF
(13) Johkoh T, et al. Idiopathic Interstitial Pneumonias: Diagnostic Accuracy of Thin-Section CT in 129 Patients. Radiology 1999; 211: 555. Free Full Text
(14) Akira M, et al. Serial computed tomographic evaluation in desquamative interstitial pneumonia. Thorax, 1997; 52: 333 - 337. Abstract
(15) Mueller-Mang C, et al. What Every Radiologist Should Know about Idiopathic Interstitial Pneumonias. RadioGraphics, 2007; 27(3): 595 - 615. Abstract
(16) Miller WT Jr. and Shah RM. Isolated Diffuse Ground-Glass Opacity in Thoracic CT: Causes and Clinical Presentations. Am. J. Roentgenol., 2005; 184(2): 613 - 622. Free Full Text
(17) Aziz AZ, et al. Interstitial Lung Disease: Effects of Thin-Section CT on Clinical Decision Making. Radiology, December 12, 2005; (2005) 2381041823. Free Full Text
(18) Gotway MB, et al. Challenges in pulmonary fibrosis · 1: Use of high resolution CT scanning of the lung for the evaluation of patients with idiopathic interstitial pneumonias. Thorax, 2007; 62: 546 - 553. Abstract
(19) Wells AU. Histopathologic Diagnosis in Diffuse Lung Disease: An Ailing Gold Standard. Am. J. Respir. Crit. Care Med., 2004; 170(8): 828 - 829. Free Full Text
(20) Valdivia E, et al. Morphology and pathogenesis of desquamative interstitial pneumonitis. Thorax, 1977; 32: 7 - 18. Abstract
(21) Fromm GB, et al. Desquamative interstitial pneumonitis. Characterization of free intraalveolar cells. Chest; 77: 552 - 554. PDF
(22) Singh G, et al. Desquamative interstitial pneumonitis. The intra-alveolar cells are macrophages. Chest; 79: 128.
(23) Inter-observer variation between pathologists in diffuse parenchymal lung disease. Thorax; 59: 500 - 505. Abstract
(24) Tubbs RR, et al. Desquamative interstitial pneumonitis. Cellular phase of fibrosing alveolitis. Chest 1977; 72: 159 - 165. PDF
(25) Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multi-disciplinary approach to diagnosis? Am J Respir Crit Care Med 2004;170(8):904–910. Free Full Text
(26) Paul K, et al. Increasing dose of methylprednisolone pulse therapy treats desquamative interstitial pneumonia in a child. Eur. Respir. J., 1999; 14: 1429 - 1432. Abstract PDF
(27) Vedal S, et al. Desquamative interstitial pneumonia. Computed tomographic findings before and after treatment with corticosteroids. Chest, 1988; 93: 215 - 217. Abstract
(28) Verleden GM, et al. Possible recurrence of desquamative interstitial pneumonitis in a single lung transplant recipient. Eur. Respir. J., 1998; 11: 971 - 974. Abstract PDF
(29) American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias . Am. J. Respir. Crit. Care Med. 165: 277-304. Free Full Text PDF
Dr. Holger Klee, 12 / 2007
zum Inhaltsverzeichnis zur Start-Seite
Liquid-Aspiration bei einem Feuerschlucker
zum Inhaltsverzeichnis zur Start-Seite
Ein 25-jähriger Mann trat regelmäßig als Feuerschlucker bei Kindergeburtstagen auf. Akzidentiell kam es wähend einer Vorführung zur Aspiration einer mittleren Menge „Terpentin“.
Der Patient stellte sich wegen leichten Allgemeinsymptomen vor. Es bestand kein Husten, keine Dyspnoe, kein Fieber. Laborwerte und Blutgasanalyse bei Aufnahme waren unauffällig.
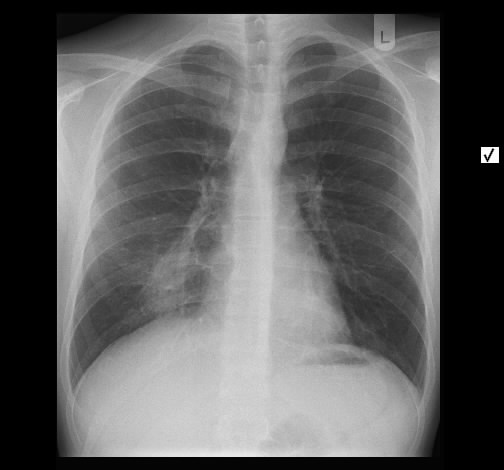
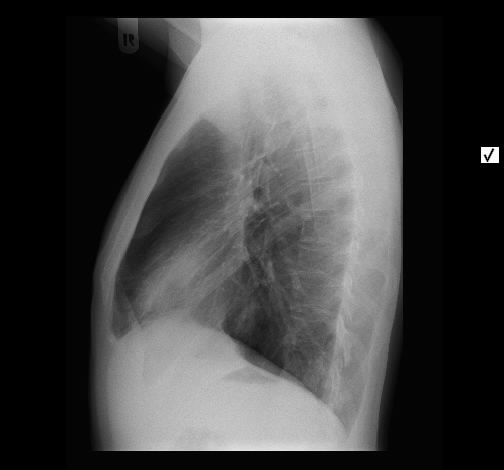
Rö-Thorax p.a.: Mittellappen-Infiltrat Rö-Thorax lateral: Mittellappen-Infiltrat
Radiologisch stellt sich ein Infiltrat im Mittellappen dar.
Professionelle und semi-professionelle Feuerschlucker verwenden als leichtflüchtige und leicht entzündliche Flüssigkeit Pyrofluidâ (1). Dabei handelt es sich um ein halogenisiertes Kohlenwasserstoffgemisch. Die genaue Zusammensetzung wird als Betriebsgeheimnis gewahrt.
Markoskopische Pathologie: Lipid-Pneumonie Histologie: exogene Lipid-Pneumonie
Die Klinik kann sehr unterschiedlich sein. Meist besteht neben leichten Allgemeinsymptomen zur geringe Zeichen einer tiefen Atemwegsaffektion. Im weiteren Verlauf können jedoch auch eine deutliche Entzündungsreaktion auftreten. (1) Der Krankheitsverlauf kann jedoch auch protrahiert von einer Entzündungsreaktion kompliziert werden. (2,3) Dennoch ist in der Regel auch bei ausgedehnten Befunden mit einer restitutio ad integrum zu rechnen. (4)
Akzidentielle Aspirationen erfolgen typischerweise in Mittellappen oder in die Unterlappen. Hier finden sich initial homogene infiltrative Verschattungen, im HR-CT mit Milchglas-Verschattungen. (5,6,7) Bei komplizierten Verlauf kann es zur Ausbildung von Pneumatozelen kommen. (6,7)
Laborparameter können initial vollständig unauffällig sein. Später kann es zu einer Erhöhung von Entzündungsparametern kommen, die als Ausdruck der entzündlichen Reaktion auf Pyrofluidâ sein können und nicht immer einer bakteriellen Superinfektion entsprechen müssen. (1,2)
In der broncho-alveolären Lavage imponieren aktivierte Makrophagen mit Lipoid-Inklusionen, sowie eine Neutrophilie und Lymphozytose. (5,9)
BAL bei Liquid-Aspiration (9)
Macrophagen
40 %
Neutrophile Granulozyten
33 %
Lymphozyten
21 %
Im weiteren Verlauf trat eine Temperaturerhöhung bis 38.5°C und ein CRP-Anstieg bis 5.7 mg/dl auf. Unter dem Verdacht einer bakteriellen Superinfektion wurde eine antibiostische Therapie mit Moxifloxacin über 7 Tage durchgeführt. Hierunter waren die Beschwerden rückläufig.
Bei zwei Kontrolluntersuchungen nach einem und vier Monaten war der radiologische Befund vollständig rückläufig. Die Lungenfunktion war altersentsprechend normal.
Lungenfunktion
VC
7.4 l
122 % des Solls
FEV-1
5.8 l
121 % des Solls
FEV1/VC
79 %
ITGV
4.6 l
131 % des Solls
R(tot)
0.08
pO2
87 mm Hg
ohne O2
pCO2
44 mm Hg
Eine weitere Nachsorge war nicht mehr notwendig. Das Hobby Feuerschlucken wurde aufgegeben.
(1) B Junge, et al. Fire eaters risk: lipoid pneumonia following aspiration of a liquid hydrocarbon mixture. Pneumologie, Sep 2002; 56: 547-9. Abstract | Full text
(2) O Karacan, I Yilmaz, and FO Eyuboglu. Fire-eaters pneumonia after aspiration of liquid paraffin. Turk J Pediatr, Jan 2006; 48: 85-8. Abstract
(3) Ewert R, et al [Aspiration of petroleum by a "fire-eater"] Pneumologie, Jun 1995; 49: 388-90. Abstract
(4) Gentina T, et al. Fire-eaters lung: seventeen cases and a review of the literature.
Medicine (Baltimore), Sep 2001; 80: 291-7. Index | Full text
(5) Kin KI, et al. Imaging of occupational lung disease. RadioGraphics, Nov 2001; 21: 1371-91. Abstract | Full text
(6) Franquet T, et al. Fire eaters pneumonia: radiographic and CT findings. J Comput Assist Tomogr, May 2000; 24: 448-50. Abstract
(7) Bankier AA, et al. Pyrofluid inhalation in "fire-eaters": sequential findings on CT.
J Thorac Imaging, Oct 1999; 14: 303-6. Abstract
(9) Burkhardt O, et al. Electron microscopic findings in BAL of a fire-eater after petroleum aspiration. Chest, Jul 2003; 124: 398-400. Abstract | Full text
Dr. Holger Klee, 7 / 2007
zum Inhaltsverzeichnis zur Start-Seite
Akute Sarkoidose mit Löfgren-Syndrom und cutaner Sarkoidose im Tatoo
zum Inhaltsverzeichnis zur Start-Seite
Sacroidosis is a fairly common disease of unknown etiology. On histological examination non-caeseous granulomas can be seen. Principially sarcoidosis can affect the whole body. But typical manifestations are found in bihilar or mediastinal lymph nodes, lung, liver, skin and (typically seen in African Americans) the heart.
An acute variant of sarkoidosis, called Löfgren-syndrome, is characterizised by bihilar lymphadenopathy, arthritis of the feet, and Erythema nodosum, which is not a manifestation of the granolomatous disease but a unspecific vascular sign.
In this case report a young woman is presented with bihilar lymphadenopathy, fever, arthritis, erythema nodosum and a skin envolvement in a tatoo. When thinking of sarcoidosis as a differential diagnosis, one should look for scars and tatoos (black tatoos predominate). This can be an easy site for histological proof.
Arthritis is often responsive to NSAR. Systemic corticoid therapy is only initiated, if there is a functional impairment of a vital or critical organ (lung, heart, eyes, CNS).
Eine 27-jährige Frau beklagte eine akute Erkrankung mit beidseitigen Gelenkbeschwerden. Insbesondere Sprunggelenke und Kniegelenke waren gerötet, überwärmt und bewegungsschmerzhaft. Die Belastbarkeit war wegen Dyspnoe merklich eingeschränkt. An beiden Unterschenkeln und Unterarmen fanden sich streckseitig schmerzhafte rötliche Hautveränderungen. Im Bereicht eines Tatoos waren drei nicht druckschmerzhafte Knötchen entstanden.
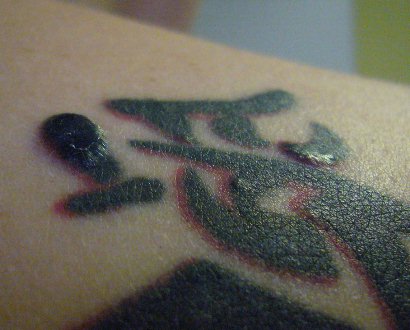
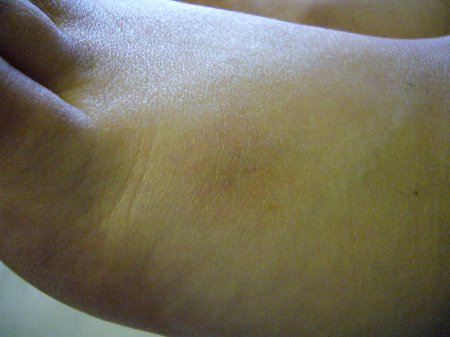
Sarkoidose im Tatoo: nicht schmerzhafte Knötchen Erythema nodosum: schmerzhafte rötlich-liquide Knoten streckseitig
Aus den klinischen Beschwerden war mit hoher Sicherheit die Diagnose eines Löfgren-Syndroms zu stellen. Es war eine Diagnosesicherung und eine Beurteilung der Differentialtherapie erforderlich.
Radiologisch stellt sich eine beidseitige hiläre und mediastinale Lymphadenopathie dar. Im HR-CT ist eine pulmonale Beteiligung nicht sicher zu sehen.
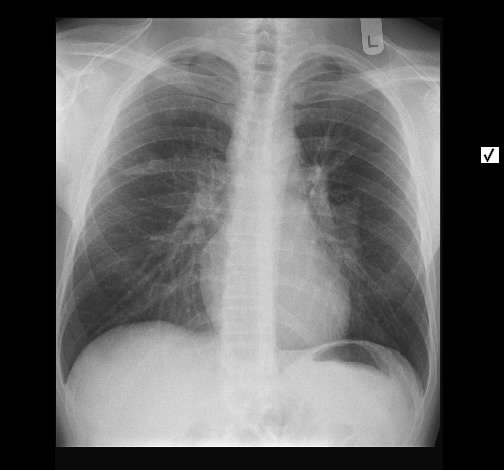
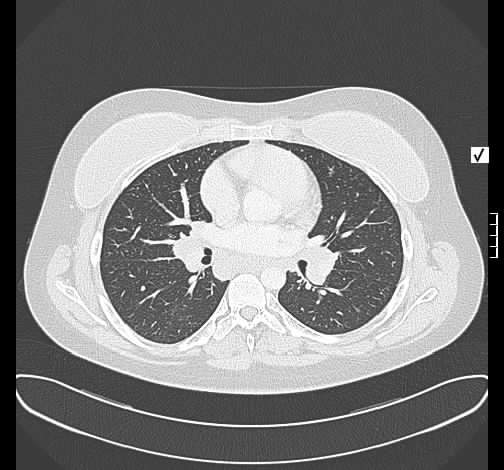
Rö-Thorax: bi-hiläre Lymphadenopathie HR- CT: mediastinale und bi-hiläre Lymphadenopathie,
keine sichere pulmonale Beteiligung
Im Labor waren Entzündungswerte erhöht, insbesondere die BSG. ACE lag im Normbereich. Hinweise auf eine Leberbeteiligung fanden sich nicht.
Labor
(Norm)
BSG
64 / 82
20 / 30
Leuko
6.5
3.5-9.8
Hb
14.2
12.0-16.0
Thr
241
140-360
Ca
2.2
2.15-2.55
Krea
0.7
0.3-0.9
GOT
11
0-35
GPT
15
0-35
GGT
22
0-40
CRP
8.4
0.0-1.0
ACE
30
8-52
Die Lungenfunktion ist unauffällig, kein Hinweis auf Restriktion, Obstruktion, Diffusionsstörung oder Atemmuskelfunktionsstörung.
Lungenfunktion
VC
4.1 l
92 % des Solls
FEV-1
3.2 l
79 % der VC
R(tot)
0.14
TLCO/VA
99 % des Solls
Pimax.
11.2
102 % des Solls
pO2
102 mm Hg
pCO2
34 mmHg
Auch in der Spiroergometrie ist eine altersentsprechend durchschnittliche Leistungsfähigkeit zu sehen. Dennoch kann natürlich eine individuelle Einschränkung bestehen, wenn zuvor ein sehr guter Trainingszustand vorlag.
Spiroergometrie
VO2max.
2052
91 % des Solls
VO2/kg
21.6
AT
1769
86 % der VO2m
AaDO2max.
23
AaDO2@AT
22
Sonographisch erschien die Milz homogen, grenzwertig groß mit 11 x 5.6 cm.
Zur Diagnosesicherung wurde eine Bronchoskopie mit BAL durchgeführt und eine Stanz-PE der Hautveränderung im Bereich des Tatoos entnommen. Auf eine periphere Lungen-PE und eine transtracheale Aspirationszytologie wurde dem Wunsch der Patientin entsprechend verzichtet. Es war ohnehin mit einer hohen diagnostischen Aussagekraft der Haut-PE zu rechnen.
Broncho-alveoläre Lavage
(Norm)
Alevolarmakroph.
88 %
86 – 98 %
Lymphozyten
10 %
0 – 10 %
Neutrophile
2 %
0 – 3 %
Eosinophile
0 %
0 – 1 %
CD4 / CD8
Das Löfgren-Syndrom bezeichnet eine akute Form der Sarkoidose mit Fieber, BSG-Erhöhung, Erythema nodosum und Polyarthritis (20-40%), meist der oberen Sprunggelenke (1,2,3).
Erythema nodosum und
bihiläre Lymphadenopathie
100 %
Fieber
92 %
Gelenkbeschwerden
89 %
Lungenbeteiligung
37 %
Hautbeteiligung
5 %
Uveitis
6 %
Parotitis
5 %
Sonstiges
8 %
Restitutio ad integrum
90 %
Betroffen sind meist junge Frauen (2,4). Zigarettenrauchen ist mit einer niedrigeren Inzidenz der Sarkoidose verbunden (5,6). Die Diagnose eines Löfgren-Syndromes stellt sich meist schon aufgrund der typischen Klinik und des Röntgenbefundes (1,2,3,7). Eine akute bilaterale Sprunggelenksarthritis bei jungen Menschen ist mit einer Sensitivität von 93 % und Spezifität von 99 % ein Löfgren-Syndrom (2).
Eine weitergehende Diagnostik mittels Transtrachealer Lymphknoten-Zytologie (8,9,10) oder BAL (11,12,13) ist selten erforderlich.
Aufgrund der guten Prognose wird in der Regel eine Therapie mit nicht-steroidalen Antirheumatika ausreichen. Eine systemisch Steroidtherapie (14,15) oder alternativen Immunsuppressiva (16,17) ist meist nicht erforderlich. Ein Progress der Erkrankung und Entwicklung einer Fibrose mit entsprechenden funktionellen und therapeutischen Konsequenzen ist nicht zu erwarten (18,19).
Neben dem typischen Löfgren-Syndrom waren bei dieser Patientin zwei unterschiedliche Manifestationen der Sarkoidose zu beobachten – Erythema nodosum und Narben-Sarkoidose im Bereich einer Tätowierung. Das klinische Spektrum der cutanen Sarkoidose ist manigfaltig. Die Häufigkeit der Hautbeteiligung wird in Studien meist um 20 - 30 % angegeben. Beide Geschlechter sind gleichermaßen betroffen (20).
Hautbeteiligung bei Sarkoidose (20)
Hautläsion
Häufigkeit
Alter
10 %
32
Lupus pernio
4 %
45
Plaques
6 %
38
Maculopapulöse Erupt.
7 %
32
Narben
1 %
40
Das Eryhthma nodusum (EN) bevorzugt die Streckseiten der Extremitäten. Es handelt sich um eine zunächst hell rote rund-ovale leicht erhabene Läsion, die sehr schmerzhaft ist. Das EN geht meist mit einer Allgemeinsymptomatik, wie Abgeschlagenheit und Fieber einher. Es ist eine unspezifische Läsion, die auch bei anderen immunologischen Erkrankungen auftritt (20,21).
Der Lupus pernio ist eine erhabene, noduläre granulomatöse Läsion, die zu einer Haut-Destruktion führt.
Sarkoidose-Plaques sind leicht erhabene, dunkel-violette Läsionen, die im Gegensatz zum EN nicht schmerzhaft sind.
plaque-förmige Sarkoidose plaque-förmige Sarkoidose
Diese Hautläsionen sind können dissiminiert in unterschiedlicher Lokalisation auftreten und sind nur leicht erhaben und nicht schmerzhaft. Sie können auch der Diagnosestellung der Sarkoidose längere Zeit vorausgehen.
makulopapulöse Sarkoidose makulopapulöse Sarkoidose
Narben können eine Prädilektionsstelle für eine Hautbeteiligung sein. Daher ist es ratsam bei dem Verdacht einer Sarkoidose systematisch Nabren abzusuchen, um möglicherweise auf einfache Art eine histologisch Sicherung zu erreichen.
Eine Sarkoidose innerhalb eines Tatoos ist ähnlich wie eine Narbensarkoidose zu werten. Tätowierungen sind Prädilektionsstellen für die Entwicklung einer Narben-Sarkoidose. Es sollten fast ausschließlich schwarzgefärbte Tatoos betroffen sein (22).
Eine Hautbeteiligung bei Sarkoidose ist häufig mit einer extrapulmonalen Organmaifestation verknüpft. Die angegebenen Zahlen sind bei der recht geringen Fallzahl nur als Anhalts zu werten.
Organbeteiligung bei cutaner Sarkoidose (n = 41)
Hautläsion
Lungen
Augen
LK
Milz
Knochen
Erythema nodosum
100 %
-
29 %
7 %
-
Lupus pernio
83 %
50 %
33 %
17 %
-
Plaques
78 %
33 %
11 %
-
11 %
Maculopapulöse Eruptionen
90 %
40 %
50 %
10 %
10 %
Narben
100 %
-
-
-
-
Bezüglich der Prognose der radiologisch darstellbaren pulmonalen Veränderungen ist das Erythema nodosum mit einer besseren Prognose für eine Restitutio ad integrum verbunden (79%) als Plaques oder Maculopapulöse Eruptionen (30%).
Von einer systemischen Steroidtherapie wurde zunächst abgesehen. Unter nicht-steroidalen Antirheumatika waren die Beschwerden bereits gut rückläufig. Die Prognose ist als sehr günstig anzusehen. Eine Resitutio ad integrum ist zu erwarten.
(1) Löfgren S. Primary pulmonary sarcoidosis: clinical course and prognosis. Acta Med Scand 1953; 145: 424-431.
(2) Paniagua O, Nugent KM. A 44-Year-Old Woman With Polyarthritis, Fever, and Hilar Adenopathy. Chest, Mar 2006; 129: 816 - 821. Free Full Text
(3) Newman, LS, Rose, CS, Maier, LA (1997) Sarcoidosis. N Engl J Med 336,1224-1234. Free Full Text
(4) Gribbin J, et al. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax, Nov 2006; 61: 980 - 985. Abstract
(5) Douglas JG, et al. Prescott, and GK Crompton. Sarcoidosis: a disorder commoner in non-smokers? Thorax, Oct 1986; 41: 787 - 791. Abstract
(6) Valeyre D, et al. Smoking and pulmonary sarcoidosis: effect of cigarette smoking on prevalence, clinical manifestations, alveolitis, and evolution of the disease. Thorax, Jul 1988; 43: 516 - 524. Abstract
(7) DeRemee RA. The roentgenographic staging of sarcoidosis. Historic and contemporary perspectives. Chest, Jan 1983; 83: 128 - 133. Free Full Text
(8) Cetinkaya E, et al. Diagnostic Value of Transbronchial Needle Aspiration by Wang 22-Gauge Cytology Needle in Intrathoracic Lymphadenopathy. Chest, Feb 2004; 125: 527 - 531. Free Full Text
(9) Trisolini R, et al. The Value of Flexible Transbronchial Needle Aspiration in the Diagnosis of Stage I Sarcoidosis. Chest, Dec 2003; 124: 2126 - 2130. Abstract
(10) Wildi SM, et al. Is endosonography guided fine needle aspiration (EUS-FNA) for sarcoidosis as good as we think? Thorax, Sep 2004; 59: 794 - 799. Abstract
(11) Drent M, et al. Bronchoalveolar lavage fluid profiles in sarcoidosis, tuberculosis, and non-Hodgkins and Hodgkins disease. An evaluation of differences. Chest, Feb 1994; 105: 514 - 519. Abstract
(12) Greco S, et al. Predictive value of BAL cellular analysis in differentiating pulmonary tuberculosis and sarcoidosis. Eur. Respir. J., August 1, 2005; 26(2): 360 - 362. Free Full Text
(13) Drent M, et al. Relationship between presentation of sarcoidosis and T lymphocyte profile. A study in bronchoalveolar lavage fluid. Chest, Sep 1993; 104: 795 - 800. Abstract
(14) Pietinalho A, et al. Oral Prednisolone Followed by Inhaled Budesonide in Newly Diagnosed Pulmonary Sarcoidosis: A Double-Blind, Placebo-Controlled Multicenter Study. Chest, Aug 1999; 116: 424 - 431. Free Full Text
(15) Gibson GJ, et al. British Thoracic Society Sarcoidosis study: effects of long term corticosteroid treatment. Thorax, Mar 1996; 51: 238 - 247. Abstract
(16) Baughman RP and Lower EE. A clinical approach to the use of methotrexate for sarcoidosis. Thorax, Aug 1999; 54: 742 - 746. Abstract
(17) WYSER CP, et al. Treatment of Progressive Pulmonary Sarcoidosis with Cyclosporin A . A Randomized Controlled Trial. Am. J. Respir. Crit. Care Med., November 1, 1997; 156(5): 1371 - 1376. Abstract
(18) Shorr AF, et al. Predicting Mortality in Patients With Sarcoidosis Awaiting Lung Transplantation. Chest, Sep 2003; 124: 922 - 928. Abstract
(19) Walker S, et al. Medium term results of lung transplantation for end stage pulmonary sarcoidosis. Thorax, Apr 1998; 53: 281 - 284. Abstract
(20) Sharma OP. Cutaneous Sarcoidosis: Clinical Features and Management. Chest, Apr 1972; 61: 320 - 325. Free Full Text
(21) MAURICE GL and TUHY JE. Erythema Nodosum and Hilar Lymphadenopathy. Chest, Mar 1954; 25: 328 - 333. PDF
(22) McGavin CR, et al. Sarcoidosis in a gecko. The Lancet 2005; 365: 1876. Full Text
Dr. Holger Klee, 10 / 07
zum Inhaltsverzeichnis zur Start-Seite
General Sherman's March to the Sea
zum Inhaltsverzeichnis zur Start-Seite
Ein 45-jähriger Mann berichtet über hartnäckigen Reizhusten, kein Auswurf, keine Dyspnoe, keine Allgemeinverschlechterung, keine Gewichtsabnahme. Der körperliche Befund ist unauffällig. Die Lungenfunktion ist normal.
Radiologisch stellen sich bds. hiläre und mediastinale Lymphknotenvergrößerungen dar. Es finden sich narbige Konsolidierungen, welche vom Mediastinum aus bis zur Pleura reichen und keine anatomischen Strukturen respektieren.
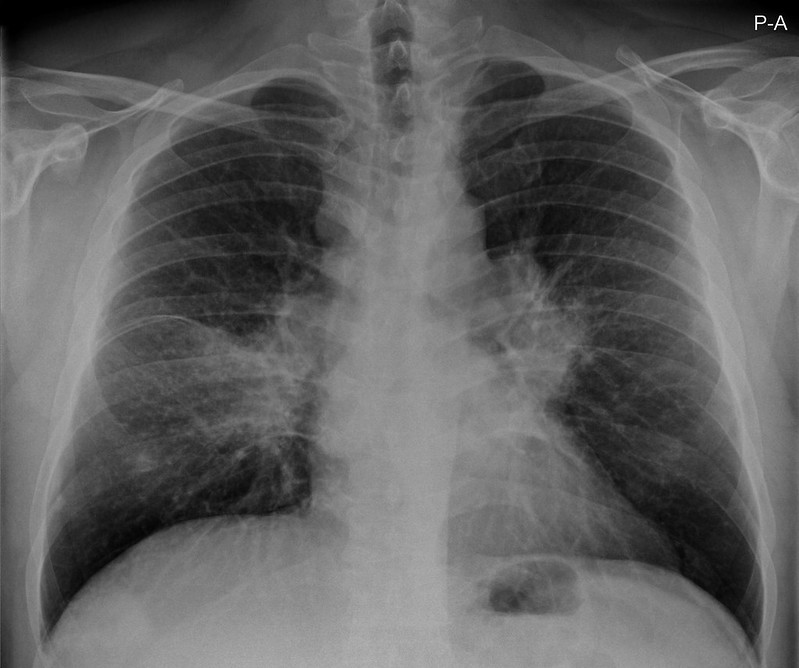
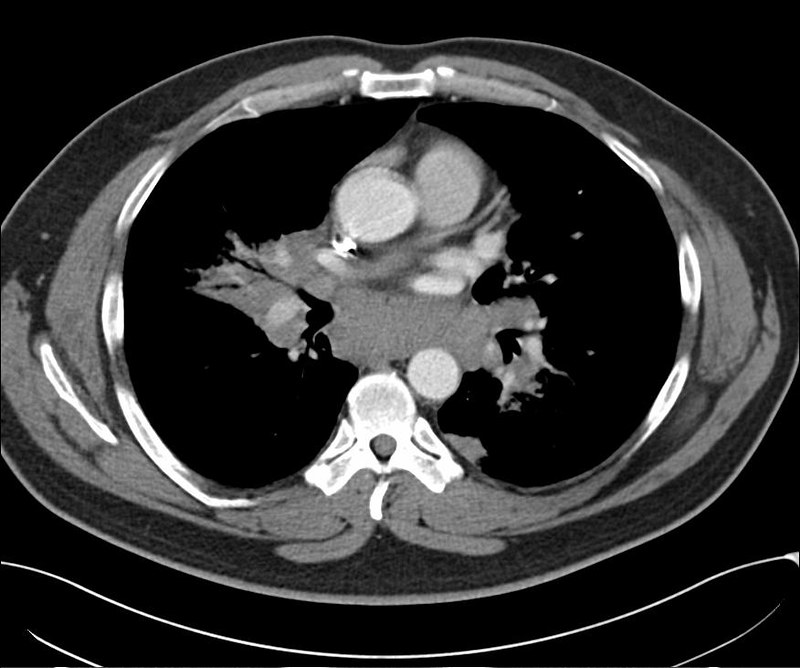
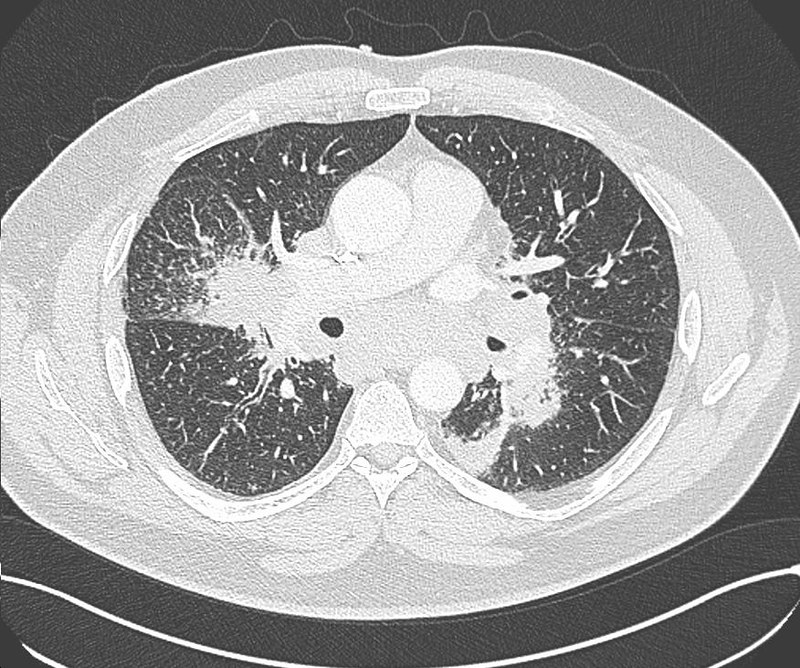
Dieses radiologische Bild wird „General Shearman’s March to the Sea“ genannt und ist typisch für eine Sarkoidose.
General Sherman kämpfte im Amerikanischen Bürgerkrieg auf Seiten der Nordstaaten.

Nach Eroberung von Atlanta zogen die Truppen der Nordstaaten marodierend durch Georgia bis an die Hafenstadt Savannah. Dabei zogen sie eine Spur der Verwüstung durch das wirtschaftliche Herz der Südstaaten. Brücken wurden zerstört, Telegrafenmasten gekappt und auf fast 500 Meilen Eisenbahnschienen demontiert, Baumwoll-verarbeitende Betriebe wurden abgebrannt.

Zwar waren bis dahin Plünderungen durch Heeresteile zur Selbstversorgung bekannt. Eine gezielte Destruktion im Hinterland des Gegners als Teil der Kriegsführung jedoch war neu. Der direkte wirtschaftliche war erheblich und wird auf (damals) $ 100 Millionen geschätzt. Unter anderem wurden 5.000 Pferde, 4.000 Maulesel, 13.000 Rinder, 9.5 Millionen Pfund Korn und 10.5 Millionen Pfund Tierfutter beschlagnahmt oder zerstört. Größer noch als der direkte wirtschaftliche Schaden war der indirekte. Durch Zerstörung der Infrastruktur waren nicht nur militärische Nachschubwegen sondern auch wirtschaftliche Transportwege zerstört. Nachdem die Baumwollverarbeitung zerstört und der wichtigste Seehafen verloren war, brach die wirtschaftliche Leistung der Südstaaten zusammen. Nach weiteren vier Monaten und einem desaströsen Feldzug durch die Carolinas kapitulierten die Südstaaten.

Eine ähnliche Schneise der Verwüstung ohne Rücksicht auf anatomische Strukturen verursacht die Sarkoidose auf dem Weg vom Mediastinum zur Pleura.
„If you see something which looks like General Shermans’s March to the Sea, think sacroid.“(1)
Literatur
(1) Gurney JW, Dobry CA: Partical Approcha to HRCT.
Dr. Holger Klee, 7 / 1012
zum Inhaltsverzeichnis zur Start-Seite
zum Inhaltsverzeichnis zur Start-Seite
Ein 41-jähriger Mann beklagte seit 3 Tagen Fieber bis 38.0°C, unproduktiven Husten und Dyspnoe. Keine Vorerkrankungen. Vom Hausarzt und einer vorbehandelnden Klinik waren unter der Differentialdiagnose einer ambulant erworbenen Pneumonie eine Antibiose mit Clarithromycin und Ceftriaxon gegeben worden ohne Effekt auf die Erkrankung.
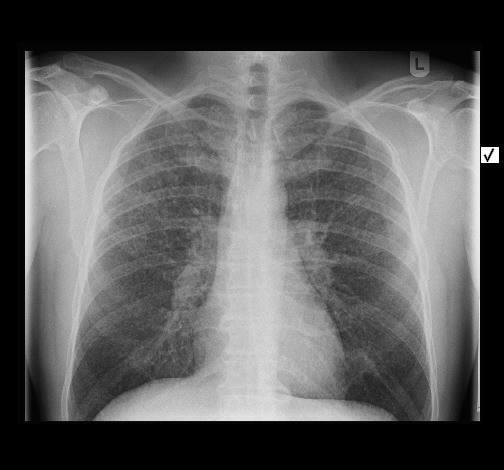
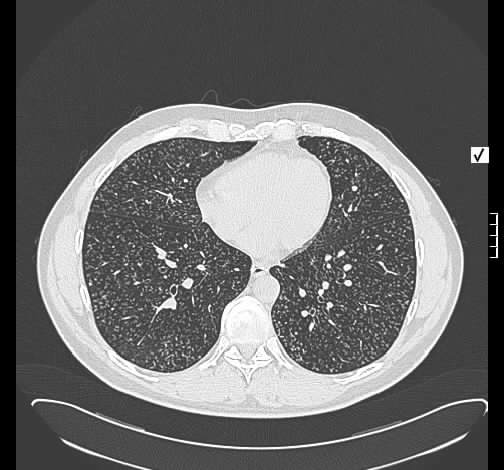
Rö-Thorax: Oberfeld-betonte feinnoduläre Zeichnugnsvermehrung Thorax-CT: feinnoduläre Zeichnungvermehrung
Radiologisch fand sich eine diffuse, oberfeld-betonte feinnoduläre Zeichnungsvermehrung.
Im Labor fand sich eine Transaminasenerhöhung. CRP war deutlich erhöht, Procalcitonin aber normal.
Labor (Norm)
Leuko
3.5 – 9.8
7.6
Hb
12.0 – 16.0
13.9
Thr
140 - 360
350
CRP
0.0 – 1.0
9.1
PCT
< 0.5
0.1
BSG
20 / 40
70 / 83
LDH
0 – 250
213
Bili
0 – 1.1
0.74
GOT
0 – 35
244
GPT
0 – 35
291
GGT
0 – 40
291
AP
35 – 105
221
Lungenfunktionell stellt sich eine leichte Restriktion und mittelgradige Obstruktion dar. In der Blutgasanalyse fand sich eine altersentsprechend mäßige Hypoxämie.
Lungenfunktion
VC
4.5 l
79 % des Solls
FEV-1
2.7 l
60 % der VC
ITGV
6.5 l
85 % des Solls
R(tot)
0.21
TLCO/VA
92 % des Solls
pO2
65 mm Hg
pCO2
37 mmHg
Es wurde eine ausgedehnte infektiologische Diferentialdiagnostik begonnen, die zu keinen richtungsweisenden Ergebnissen führte.
Infektiologische Differentialdiagnostik
Chlamydien-AK
negativ
Legionellen-AK
negativ
Mycoplasen KBR
negativ
RSV-AK
negativ
Hepatits A-B-C
negativ
Säurefeste Stäbchen (BAL)
negativ
Säurefeste Stäbchen PCR
negativ
Pneumocystic jiroveci (BAL)
negativ
In der BAL stellte sich eine deutliche Lymphzytose und eine leichte Eosinophilie. Der CD4/CD8-Quotient war normal.
BAL (Norm)
Gesamtzellzahl
4 –10 Mill.
4 Mill.
Lymphzyten
0 – 10 %
31 %
Neutrophile
0 – 3 %
3 %
Eosinophile
0 – 1 %
14 %
CD4/CD8-Quotient
2
Eine periphere Lungen-PE zeigte ödematöse und entzündlich infiltrierte Alveolarspeten und intraalveoläre Proliferate aus fibroblastären Zellen. Keine Fibrose und kein Nachweis von säurefesten Stäbchen oder von Granulomen. Zusammenfassend war die Histologie vereinbar mit einer BOOP-Reaktion.
Die Bronchiolitis obliterans organisierende Pneumonie (BOOP) ist eine klinisch, radilogisch und histologisch charakterisierte interstitielle Lungenerkrankung. Im anglo-amerikanischen Sprachraum wird der Ausdruck Cryptogen organisierende Pneumonie COP bevorzugt, was dem regelmäßig anzutreffenden histologischen Merkmal entspricht. (1,2,35,36)
Man nimmt eine immunologische Reaktion auf eine Vielzahl unterschiedlicher Noxen an. Oft ist die Ätiologie nicht zu klären, so dass von einer idiopathischen BOOP gesprochen werden muß. (41)
Einteilung der BOOP nach Ätiologie (3)
Idiopathische BOOP
Rapid progressive BOOP (4)
Fokal noduläre BOOP
Post-Infektiös
Chlamydien, Coxiellen, Legionellen Mycoplasmen, Nocardien, Pseudomonas
Serratia, Staphylokokken, Streptokokken
Herpes simplex, HIV, Influenza, Parainfl.
Plasmodium vivax (5)
Cryptococcus, Penicillum, PCP
Medikamente (Auswahl) (6)
Amphotericin B, Cephalosporine
Minocilin, Nirofurantoin (7,8)
Sulfamethoypyridazin, Sulfasalazin
Bleomycin, Gold, Amiodaron
Phenytoin, Carbamazepin, Ticlopidin
Rheumatologische Erkrankungen
Lupus erythematosus (9)
Rheumatoide Arthritis
Sjögren-Syndrom, Sweet Syndrom
Sklerodermie, Bechterew, Behcet
Polymyalgie, Polymyositis
Dermatomyositis, Panarteritis nod. (10)
Immunologische Erkrankungen
CVIDS, Kryoglobulinämie
Transplantation (11)
Bestrahlung (29)
Umwelttoxine
Stoff-Farben, Rauch-Exposition
Verschiedene
Colitis ulcerosa / M. Crohn
Lymphom, Carciom, MDS
Thyreoiditis, Leberzirrhose, PCB, ACVB
Die BOOP wurde als Krankheitsendität erstmal 1985 beschrieben. (1) Seitdem ist die Diagnose weltweit zunehmend häufig gestellt worden. Valide epidemiologische Daten zur Inzidenz oder Prävalenz liegen nicht vor. (32) Kein Geschlecht ist bevorzugt betroffen. Nichtraucher sind unter den Patienten häufiger im Verhältnis 2:1. Das durchschnittliche Alter bei Diagnosestellung beträgt 55 Jahre. (12,2)
Klinisch imponiert meist ein unspezifisches respiratorisches Syndrom mit Fieber, unproduktivem Husten, Dyspnoe und Gewichtsverlust. Hämoptysen (28), Thoraxschmerzen, Arthralgien und Nachtschweiß sind selten. (13,31) In der weit überwiegenden Mehrzahl der Fälle entwickelt sich das Krankheitsbild über mehrere Wochen, evtl. im Anschluß an einen tiefen Atemwegsinfekt. Eine Vorgeschichte mit mehrfachen, erfolglosen Therapieversuchen mit unterschiedlichen Antibiotika ist typisch. (39)
Sehr selten ist ein hoch akutes, lebensbedrohliches Krankheitsbild, das klinisch von einem ARDS nur schwer zu unterscheiden ist. (13,4)
Klinik und Befunde der BOOP (14)
Husten
76 %
Fieber
53 %
Dyspnoe
47 %
Sputum
9 %
Rasselgeräusche
79 %
Trommelschlegelfinger
3 %
CRP-Erhöhung
79 %
Lymphozytose
38 %
Radiologisch imponieren überwiegend fleckige Infiltrate, die eher in den Unterfeldern zu finden sind. Seltener treten subpleurale Konsolidierungen auf. Die Infiltrate können im Verlauf wechseln und auch in der Gegenseite auftreten. Feinnoduläre oder retikuläre Infiltrate - wie in dem beschreibenen Fall - sind selten.
Radilogische Befunde der BOOP (14)
Fleckige Infiltrate
68 %
Retikuläre Infiltrate
6 %
Lineare Verschattungen
15 %
Oberfelder
24 %
Mittelfelder
26 %
Unterfelder
65%
Den häufigeren radiologischen Befund von bilateralen dreiecksförmigen Konsolidierungen beschreibt der untenstehende Case Report.
Nach Corier (1989) werden drei Gruppen von radiologischen Befunden beschrieben, die sich auch im klinischen Verlauf unterscheiden. (15,38)
Radilogische Gruppierung der BOOP (15)
Multiple alveoläre Infiltrate
gutes Ansprechen auf Steroide, Rezidivneigung
Singuläres alveoläres Infiltrat oder Herdbefund
durch Resektion heilbar (30)
Interstitielle Infiltrate
meist deutliche Dyspnoe,
evtl. schlechte Prognose
Rö-Thorax: bds. alveoläre Infiltrate (2)
Im Thorax-CT stellen sich sich subpleurale oder peribronchiale Infiltrate dar. In dichteren Infiltraten finden sich Aero-Bronchogramme, in weniger dichten Infiltraten kommen Gefäße zur Darstellung. (16,34) Kleine peri-bronchovasculäre Noduli (10 – 50 %) und Milchglas-Verschattungen (60 – 100 %) können vorkommen. (2) Multiple kleine Noduli bestimmen in 30 % der Patienten das radiologische Bild. Noduläre Veränderungen sind bei immunkomprimierten Patienten häufiger. (17) Pleuraergüsse sind selten. In einer kleinen Gruppe von Patienten (15 %) können auch größere Knoten oder Konsolidierugen beobachtet werden. (2,16,40)
CT-Befunde der BOOP (18)
Milchglas-Verschattungen 90 – 100 %
fleckige Konsolidierungen 83 – 91 %
bilateral und peripher betont, evtl. dorsal
dreiecksförmig
peribronchiale Infiltrate in Richtung Pleura
subpleurale Verdichtung ohne anatom. Regel
selten retikuläre oder größere fokale Läsionen
Thorax-CT: peripheres dreiecksförmiges Infiltrat mit Aerobronchogramm (2)
Thorax-CT: periphere dreiecksförmige Infiltrate und Konsolidierungen mit Aerobronchogrammen (38)
Thorax-CT: peribronchovaskuläres Milchglas und subpleurale Konsoludierung (39)
Lungenfunktionell kann eine leicht- bis mittelgradige Restriktion vorliegen (15,12). Eine Obstruktion steht wahrscheinlich im Zusammenhang mit einer etwaigen Nikotinanamnese. (15) Der CO-Transfer-faktor ist meist mäßig erniedrigt. (2) Eine milde bis selten auch schwere belastungsinduzierbare Hypoxämie ist Folge eines Rechts-Links-Shunts. (15,13,12)
Die Diagnosestellung erfolgt in der Zusammenschau aus anamnestischen, klinischen und radiologischen Befunden. Sie wird unterstützt durch Befunde der Histologie und broncho-alvolären Lavage. Eine bronchoskopisch gewonnen periphere PE kann häufig differentialdiagnostisch ausreichend sein, so dass häufig eine offene chirurgische PE nicht zwingend erforderlich wird. (19)
Histologisch zeichnet sich die BOOP durch eine Oraganisationspfröpfe aus myxoidem, fibroblastenreichem Bindegewebe in den Alveolen (organisierende Pneumonie) und Bindegewebsproliferationen in den Bronchiolen (Bronchiolitis obliterans) aus. Dabei kann die zweite Komponente fehlen. (18,20,21)
Typ. histologische Befunde bei BOOP (2)
intraluminale organisierende Fibrose
Fleckige Verteilung
Lungenarchitektur erhalten
zeitlich homogen
milde chronische interstitielle Entzündung
Keine Fibrose, Granulome, Neurophile, Abszesse, Nekrosen, hyaline Membranen, Eosinophile, Vaskulitis
Histologie: BO und COP (3) Histologie: BO und COP (33)
Histologie: frühes Stadium (39) Histologie: spätes Stadium (39)
Eine BOOP-Reaktion ist keine spezifische histologische zu charakterisierende Endität. Sie kommt bei einer Reihe von immunologische Reaktionen vor. Die histologische Differentialdiagnose zur BOOP ist breit. (22)
In der BAL ist eine mäßige Lymphozytose mit erniedrigtem CD4/CD8-Quotient zu erwarten. (2,23) Eine leichte Eosinophilie in der BAL ist bei BOOP häufig anzutreffen. Eine ausgeprägte Eosinophilie (> 20 %) sollte an die Differentialdiagnose einer Chronisch Eosinophilen Pneumonie denken lassen. (12,24) Eine Lymphozytose > 20 % spricht gegen eine idiopathische pulmonale Fibrose (IPF / UIP), Leu-7+-Lymphozyten > 15 % sind bei einer Exogen Allergischen Alveolitis zu erwarten. (24,37)
BAL bei BOOP (24)
Lymphozytose > 15 %
100 %
Neutrophilie > 3 %
57 %
Eosinophilie > 1 %
86 %
Plasmazellen
50 %
Schaum-Makrophagen > 20 %
34 %
CD4/CD8-Quatient < 1.0
93 %
Leu-7-Lymphozyten < 15 %
100 %
HLA-DR+-Lympho > 5 %
57 %
BAL in der Differentialdiagnose von ILD (37)
Empfohlen wird eine systemische Steroidtherapie, beginnend mit 40 mg pro Tag und Reduktion alle 4 Wochen um 10 mg. Eine Erhaltungsdosis von 10 mg für insgesamt 12 Monate wird vorgeschlagen. (12)
Die Wertigkeit von Clarithromycin als Therapiealternative ist zurzeit noch nicht abschließend beurteilbar. (33)
Unter einer systemischen Steroidthearpie kann mit einer raschen klinischen Besserung gerechnet werden. Die radiologischen Veränderungen verschwinden im Durchschnitt nach 10 Wochen vollständig. (12)
In einem Drittel bis der Häfte der Patienten tritt ein Relaps auf, oft auch wiederholt. (25) Rezidiverkrankungen sprechen ebenso gut und mit gleich guter Prognose auf eine erneute Steroidtherapie an. Risikofaktoren für einen Relpas sind verzögerter Therapiebeginn, eine leichte Cholestase, eine schwere initiale Hypoxämie und möglicherweise auch eine Hypalbuminämie. (27)
Häufigkeit der Rezidive (26) BAL und Lungenfunktion als Prognoseparameter für ein Rezidiv (26)
Wegen möglicher Nebenwirkungen einer längerfristigen systemischen Steroidtherapie wird dennoch empfohlen, die Steroiddosis möglichst dem Standard-Regime rasch zu reduzieren. (26,27)
Die Prognose quo ad vitam ist in aller Regel gut. In einer japanischen Studie wurde eine Letalität von 3 % (2/34) gefunden. (14) Nur sehr selten tritt ein fulminanter, lebensbedrohlicher Verlauf auf, der dann klinisch von einem ARDS anderer Genese nicht zu unterscheiden ist. (4)
Es wurde eine systemische Steroidtherapie beginnend mit 40 mg Prednisolon-Äquivalent pro Tag begonnen. Eine klinische Besserung war nach einer Woche zu sehen. Die radiologischen und lungenfunktionellen Veränderungen waren nach zwei Wochen rückläufig. Nach stufenweiser Reduktion wurde eine systemische Steroidtherapie mit 10 mg Prednisolon-Äquivalent über insgesamt 12 Monate fortgeführt. Während einer dreijährigen Nachbeobachtung ist kein Rezidiv aufgetreten.
(1) Epler GR, Colby TV, McLoud TC, Carrington CB, Gaensler EA. Bronchiolitis obliterans organizing pneumonia. N Engl J Med. 1985;312:152-158. Abstract
(2) Travis WD, King TE, Bateman ED, et al. ATS/ERS international multidisciplinary consensus classification of idiopathic interstitial pneumonias. General principles and recommendations. Am J Respir Crit Care Med 2002;165:277–304. Free Full Text
(3) Epler GR. Bronchiolitis obliterans organizing pneumonia. Arch Intern Med, Jan 2001; 161: 158-64. Free Full Text
(4) Nizami IY, et al. Idiopathic Bronchiolitis Obliterans With Organizing Pneumonia: An Acute and Life-Threatening Syndrome. Chest, Jul 1995; 108: 271 - 277. PDF
(5) Yale SH, et al. Bronchiolitis obliterans organizing pneumonia caused by Plasmodium vivax malaria. Chest, Oct 1993; 104: 1294 - 1296. PDF
(6) Rosenow EC, 3d, et al. Drug-induced pulmonary disease. An update. Chest, Jul 1992; 102: 239 - 250. PDF
(7) Fawcett W an Ibrahim NBN. BOOP associated with nitrofurantoin. Thorax, Feb 2001; 56: 161. Abstract
(8) Cameron RJ, et al. Bronchiolitis obliterans organising pneumonia associated with the use of nitrofurantoin. Thorax, Mar 2000; 55: 249 - 251. Abstract
(9) Gammon RB, et al. Bronchiolitis obliterans organizing pneumonia associated with systemic lupus erythematosus. Chest, Oct 1992; 102: 1171 - 1174. Abstract
(10) Robinson BW and Sterrett G. Bronchiolitis obliterans associated with polyarteritis nodosa. Chest, Jul 1992; 102: 309 - 311. PDF
(11) Chaparro C, et al. Bronchiolitis Obliterans Organizing Pneumonia (BOOP) in Lung Transplant Recipients. Chest, Nov 1996; 110: 1150 - 1154. Abstract
(12) U Costabel, et al. BOOP in Europe. Chest, Jul 1992; 102: 14S - 20S. PDF
(13) Cordier JF. Organising pneumonia. Thorax. 2000;55:318-328. Free Full Text
(14) Izumi T, et al. Bronchiolitis obliterans organizing pneumonia. Clinical features and differential diagnosis. Chest, Sep 1992; 102: 715 - 719. PDF
(15) Cordier JF, Loire R, Brune J. Idiopathic bronchiolitis obliterans organizing pneumonia. Definition of characteristic clinical profiles in a series of 16 patients. Chest 1989; 96: 999-1004. Abstract PDF
(16) Nishimura K and Itoh H. High-resolution computed tomographic features of bronchiolitis obliterans organizing pneumonia. Chest, Jul 1992; 102: 26S - 31S. PDF
(17) Lee KS, Kullnig P, Hartman TE, Muller NL. Cryptogenic organizing pneumonia: CT findings in 43 patients. Am J Roentgenol 1994; 162: 543-546. Abstract PDF
(18) Cordiere JF. Cryptogenic organising pneumonia. Eur. Respir. J., Aug 2006; 28: 422 - 446. Abstract
(19) Azzam ZS, et al. Bronchiolitis obliterans organizing pneumonia. Diagnosis by transbronchial biopsy. Chest, Dec 1993; 104: 1899 - 1901. ODF
(20) Epler GR. Bronchiolitis obliterans organizing pneumonia: definition and clinical features. Chest, Jul 1992; 102: 2S - 6S. PDF
(21) Epler GR. Bronchiolitis obliterans organizing pneumonia. Arch Intern Med, Jan 2001; 161: 158-64. Free Full Text
(22) Kitaichi M. Differential diagnosis of bronchiolitis obliterans organizing pneumonia
Chest, Jul 1992; 102: 44S - 49S. PDF
(23) Nagai S, et al. Bronchoalveolar lavage cell findings in patients with BOOP and related diseases. Chest, Jul 1992; 102: 32S - 37S. PDF
(24) Costabel J, Teschler H, and Guzman K. Bronchiolitis obliterans organizing pneumonia (BOOP): the cytological and immunocytological profile of bronchoalveolar lavage. Eur. Respir. J., Jul 1992; 5: 791 - 797. Abstract
(25) Epler GR. Bronchiolitis Obliterans Organizing Pneumonia. Arch Intern Med, January 22, 2001; 161(2): 158 - 164.
(26) Lazor R, et al. Cryptogenic Organizing Pneumonia . Characteristics of Relapses in a Series of 48 Patients. Am. J. Respir. Crit. Care Med. 162: 571-577. Free Full Text
(27) Watanabe K, et al. Factors Related to the Relapse of Bronchiolitis Obliterans Organizing Pneumonia. Chest, Dec 1998; 114: 1599 - 1606. PDF
(28) Mroz BJ, et al. Hemoptysis as the Presenting Symptom in Bronchiolitis Obliterans Organizing Pneumonia. Chest, Jun 1997; 111: 1775 - 1778. Abstract
(29) Lazor R, et al. Bronchiolitis Obliterans Organizing Pneumonia Syndrome Primed by Radiation Therapy to the Breast. Am. J. Respir. Crit. Care Med. 158: 1929-1935. Free Full Text
(30) Akira M, Yamamoto S, Sakatani M. Bronchiolitis obliterans organizing pneumonia manifesting as multiple large nodules or masses. Am J Roentgenol 1998; 170: 291-295. Abstract PDF
(31) Yamamoto M, et al. Clinical features of BOOP in Japan. Chest, Jul 1992; 102: 21S - 25S. PDF
(32) Cordier J.-F.Rare diseases bullet 8: Organising pneumonia. Thorax, April 1, 2000; 55(4): 318 - 328. Abstract
(33) Stover DE and Mangino D. Macrolides: A Treatment Alternative for Bronchiolitis Obliterans Organizing Pneumonia? Chest, Nov 2005; 128: 3611 - 3617. Free Full Text
(34) Nishimura K, et al. The diagnostic accuracy of high-resolution computed tomography in diffuse infiltrative lung diseases. Chest, Oct 1993; 104: 1149 - 1155. PDF
(35) Geddes DM. BOOP and COP. Thorax, Aug 1991; 46: 545 - 547. Abstract
(36) Cordier JF. Cryptogenic organising pneumonia. Eur. Respir. J., August 1, 2006; 28(2): 422 - 446. Abstract
(37) Welker L, et al. Predictive value of BAL cell differentials in the diagnosis of interstitial lung diseases. Eur. Respir. J., Dec 2004; 24: 1000 - 1006. Free Full Text
Tbl. BAL
https://erj.ersjournals.com/cgi/content-nw/full/24/6/1000/T2
(38) Lynch DA, et al. Idiopathic Interstitial Pneumonias: CT Features. Radiology 2005; 236: 10-21. Free Full Text
(39) Wittram C, et al. CT-Histologic Correlation of the ATS/ERS 2002 Classification of Idiopathic Interstitial Pneumonias. RadioGraphics 2003; 23: 1057-1071. Free Full Text
(40) Johkoh T, et al. Idiopathic Interstitial Pneumonias: Diagnostic Accuracy of Thin-Section CT in 129 Patients Radiology 1999; 211: 555. Abstract Free Full Text
(41) Cordier, J-F. Cryptogenic organising pneumonia. Eur. Respir. J., Aug 2006; 28: 422 - 446. Abstract
Dr. Holger Klee, 1 / 2008
zum Inhaltsverzeichnis zur Start-Seite
zum Inhaltsverzeichnis zur Start-Seite
Ein 60-jähriger Mann berichtete seit drei Monaten anhaltenden unproduktiven Reizhusten, subfebrile Temperaturerhöhung und allgemeine Abgeschlagenheit. Unter dem Verdacht einer Pneumonie waren drei verschiedene Antbiotika gegeben worden. Sieben Jahre zuvor war wegen einem Oesophaguscarcinom eine Resektion und Coloninterposition durchgeführt worden. Bislang kein Rezidivhinweis.
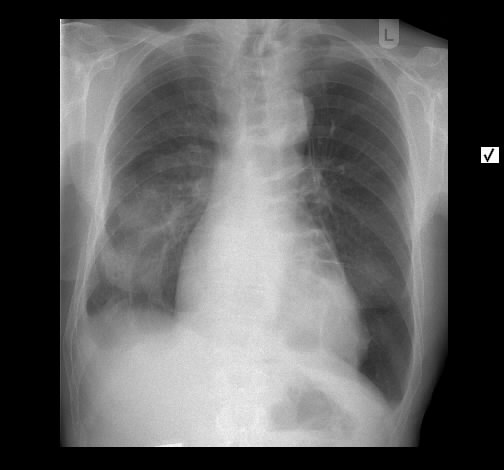
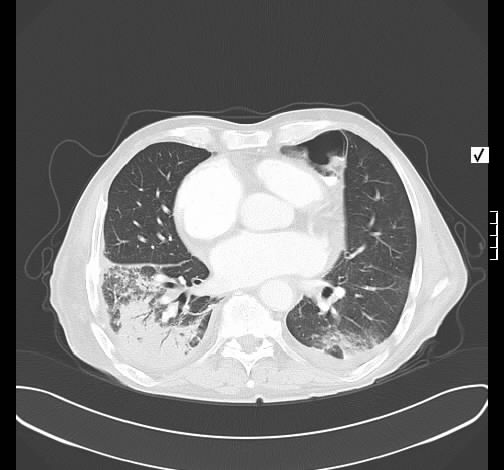
Rö-Thorax: fleckige Infiltration in den Unterlappen rechts betont Thorax-CT: periphere dreiecksförmige Konsolidierung mit
Nebenbefundlich links präkordiales Coloninterponat Aerobronchogramm und diskreten Pleuraergüssen bds.
Die Anamnese mit fehlendem Ansprechen auf Antibiosen, die Klinik und fehlende laborchemische Entzündungsparameter sprechen gegen eine bakterielle Pneumonie. Eine Tuberkulose ist denkbar. Ein Tuberkulin-Hauttest war negativ und Sputen ohne mikroskopischen Nachweis von säurefesten Stäbchen.
Differentialdiagnostisch kommte gerade in Anbetracht des CT-radiologischen Befundes eine Bronchiolitis obliterans organisierende Pneumonie BOOP oder eine chronische eosinophile Pneumonie CEP in Betracht.
Ich verweise auf die Differentialdiagnostik gegenüber einer Chronischen Eosinophilen Pneumonie (CEP) weiter oben.
Differentialdiagnose zwischen CEP und BOOP (1)
flaue Infiltrate peripher
dichtes dreiecksförmiges Infiltrat
Mittel-Oberfeld
Mittel-Unterfeld
selten Aerobronchgramm
häufigAerobronchogramm
verdickte Septen 72 %
verdickte Septen 40 %
Noduli 5 %
Noduli 32 %
retikuläres Muster 9 %
retikuläres Muster 45%
Bronchialerweiterung 26 %
Bronchialerweiterung 58%
peribronchial 9 %
peribronchial 29 %
Eosinophilie>20 %
buntes Zellbild
keine Fibrose
fibroblastenreich
Schaumzellen selten
Schaumzellen häufig
BO selten
BO häufig
Für eine BOOP spricht insbesondere das typisch dreiecks-förmige dichte Infiltrat mit Aerobronchogramm und die Betonung von Unter-Mittelfeldern.
Radilogische Befunde der BOOP (2)
Fleckige Infiltrate
68 %
Retikuläre Infiltrate
6 %
Lineare Verschattungen
15 %
Oberfelder
24 %
Mittelfelder
26 %
Unterfelder
65%
CT-Befunde der BOOP (3)
Milchglas-Verschattungen 90 – 100 %
fleckige Konsolidierungen 83 – 91 %
bilateral und peripher betont, evtl. dorsal
dreiecksförmig
peribronchiale Infiltrate in Richtung Pleura
subpleurale Verdichtung ohne anatom. Regel
selten retikuläre oder größere fokale Läsionen
Zur Beschreibung der BOOP verweise ich weiterhin auf den obenstehenden Case Report.
Dem Wunsch des Patienten entsprechend wurde auf eine weitere Diagnostik verzichtet und ein Therapieversuch mit einem systemischen Steroiden begonnen in einer Initialdosis von 40 mg Prednisolon-Äquivalent pro Tag. Nach vier Wochen war das Infiltrat vollständig rückläufig. Dies unterstützte die Arbeitsdiagnose einer BOOP.
Die Dosis wurde nach zwei Monaten stufenweise auf 10 mg Prednisolon-Äquivalent reduziert. Nach einer Therapiedauer von insgesamt 9 Monaten konnte die Medikation beendet werden. In einer 5-jährigen Nachbeobachtung ist kein Rezidiv aufgetreten.
(1) Arakawa H, et al Bronchiolitis Obliterans with Organizing Pneumonia Versus Chronic Eosinophilic Pneumonia - High-Resolution CT Findings in 81 Patients AJR 2001; 176:1053-1058. Free Full Text Abstract
(2) Izumi T, et al. Bronchiolitis obliterans organizing pneumonia. Clinical features and differential diagnosis. Chest, Sep 1992; 102: 715 - 719. PDF
(3) Cordier JF, Loire R, Brune J. Idiopathic bronchiolitis obliterans organizing pneumonia. Definition of characteristic clinical profiles in a series of 16 patients. Chest 1989; 96: 999-1004. Abstract PDF
Dr. Holger Klee, 1 / 2008
zum Inhaltsverzeichnis zur Start-Seite
Sklerodermie (CREST-Syndrom) mit Lungenbeteiligung und pulmonaler Hypertonie
zum Inhaltsverzeichnis zur Start-Seite
Ein 73-jähriger Patient wurde seit ca. 15 Jahren wegen derben Hautveränderungen rheumatologisch betreut. Es wurde in einer spezialisierten rheumatologischen Abteilung die Diagnose einer Sklerodermie gestellt.
Wegen geringgradiger Belastungsdyspnoe wurde der Patient pneumologisch vorgestellt und eine pulmonale Beteiligung dokumentiert. Nach weiteren 5 Jahren sind klinische Beschwerden und radiologischer Befund unverändert.
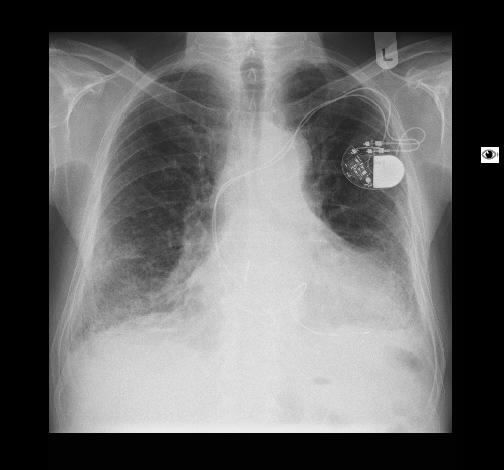
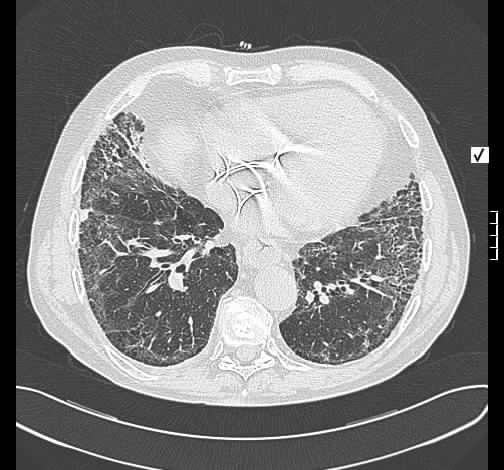
Rö-Thorax: interstitielle Zeichungsvermehrung bds. basal, DDD-SM Thorax-CT: basal retikuläre Zeichnungsvermehrung, diskretes honey combing
Das radiologische Bild läßt sich formal als UIP-ähnlich beschreiben.
Weitere Amanese und Krankheitsverlauf
Therapieversuche mit sytemischen Steroiden, Methotrexat und Cyclophosphamid konnten den Krankheitsverlauf nicht erkennbar beeinflussen. Die Verträglichkeit der immunmodulatorischen Therapie war schlecht gewesen. Zuletzt wrude ein Therapie mit Hydrochloroquin eingeleitet.
Im Krankheitsverlauf traten derbe Hautindurationen auf mit ausgestanzten, areaktiv imponierenden Ulzera. Radiologisch stellen sich gelenknahe subkutane Verkalkungen dar.
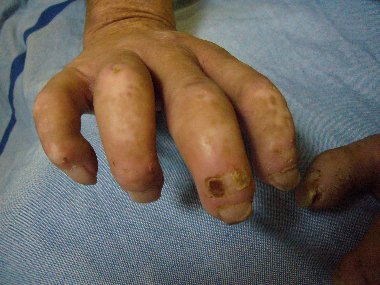
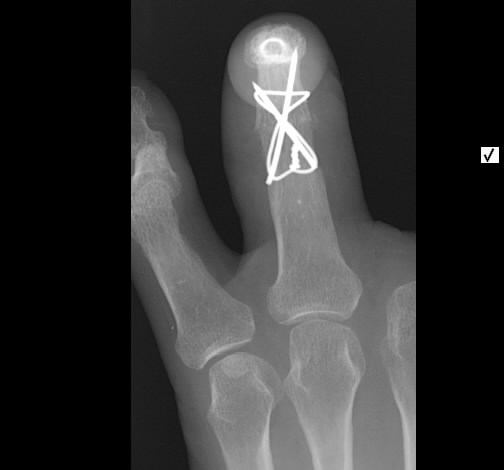
Foto: derbe Hautinduration, gelenknahe Ulzera Rö: gelenknahe subkutane Verkalkungen
Nach langjährigem Raynaud-Syndrom traten akrale Nekrosen an den Fingerspitzen und an der Nasenspitze auf.
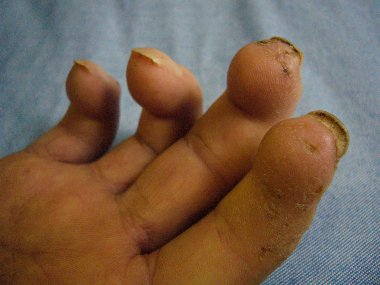
Foto: akrale Nekrosen
Wegen Schluckbeschwerden waren zweimalig Oesophagus-Bougierungen notwendig geworden. Im Thorax-CT stellt sich dennoch eine Erweiterung des Oesophgus dar.
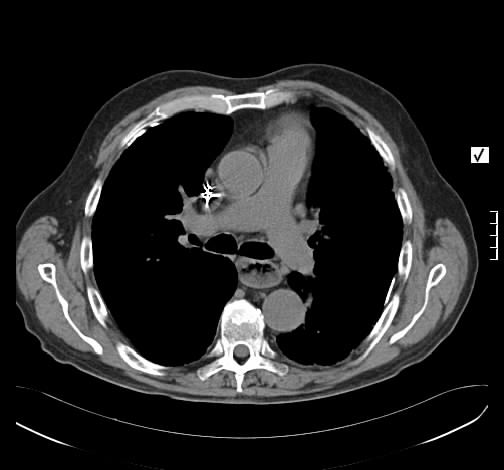
Thorax-CT: erweiterer Oesophagus
Insbesondere die Finger imponieren derb, verdickt und kaum beweglich.
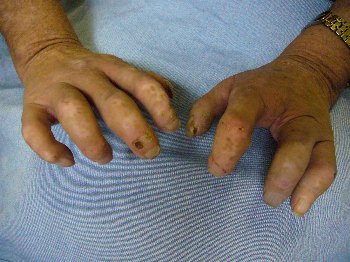
Foto: Sklerodaclylie
Im Bereich der Wangen sind Teleangiektasien zu sehen. Weiterhin stellt sich ein Tabaksbeutelmund und eine Vitiligo der Kopfhaut dar.
Foto: Teleangiektasien, Tabaksbeutelmund, Vitiligo
Diagnosestellung
Zusammenfassend ergab sich aus den oben angeführten Befunden das klinische Vollbild eines CREST-Syndromes.
CREST-Syndrom (5,8)
Calcinosis cutis
Raynaud-Syndrom
Esophagus-Motilitätsstörungen
Sklerodactylie
Teleangiektasien
Die Diagnose wurde durch das Antikörpermuster gestützt.
Antikörpermuster
ANA
mit gesprenkeltem Muster
1 : 5120
Scl-70-AK-Ratio
6.8
Dabei entsprechen ANA mit gesprenkeltem Floureszenzmuster Anti-Zentromer-Antikörpern, die für ein CREST-Syndrom typisch sind.
Lungenfunktion
In der Lungenfunktion zeigt sich eine mittelgradige Restriktion und leichte Diffusionsstörung.
Lungenfunktion
VC
2.3 l
60 % des Solls
FEV-1
1.9 l
82 % der VC
ITGV
2.6 l
85 % des Solls
R(tot)
0.11
TLCO/VA
76 % des Solls
pO2
88 mm Hg
pCO2
36 mmHg
Spiroergometrie
Spiroergometrisch ist die Leistungsfähigkeit mittel- bis schwergradig erniedrigt. Eine relevante Diffusionsstörung oder Verteilungsstörung kann bei fast normaler AaDO2 ausgeschlossen werden. Ein erhöhtes Atemäqzuivalent für CO2 spricht für eine leicht- bis mittelgradige pulmonale Hypertonie.
Spiroergometrie
VO2max.
1.116 l/min
65 % des Solls
AaDO2max.
30 mmHg
leicht erhöht
VE/VCO2
50
erhöht
PET CO2
27 mmHg
normal
Echokardiographie
Echokardiographisch bestätigte sich der Verdacht einer leicht- bis mittelgradigen pulmonalen Hypertonie. Der PAPs wurde mit 35 – 40 mmHg gemessen.
Definition Sklerodermie
Die Sklerodermie stellt ein Spektrum vermutlich autoimmun-bedingter Erkrankungen dar, die durch Autoimmunphänomephe, Vaskulopathie und progressive interstitielle und perivaskuläre Fibrosierung gekennzeichnet sind. Neben der Krankheitsbezeichung Sklerodermie ist auch Systemische Sklerose üblich.
Histologie: kutane Sklerodermie
Man unterscheidet nach der Ausprägung des Hautbefalls zwei unterschiedliche Pole des Erkrankungsspektrum. Neben einem diffusem kutanen Befall, der häufiger mit einer systemischen Organbeteiligung korreliert ist, kommt auch eine limitierete / zirkumskripte kutane Manifestation vor. (1)
Epidemiologie
Die Sklerodermie ist eine insgesamt seltene Erkrankung mit einer Prävalenz von 242 / 1.000.000 Einwohnern (0.024 %). Es muß mit einer Inzidenz von ca. 19 Neuerkrankungen pro 1.000.000 Erwachserer gerechnet werden. Dabei sind Frauen und Afro-Amerikaner häufiger betroffen. (2)
Lungenbeteiligung bei Sklerodermie
Komplikationen des Respirationstraktes bei Sklerodermie umfassen neben möglichen Aspirationen oder extrathorakeler Restriktion durch ausgedehnte Hautbeteiligung (3) insbesondere interstitiell-fibrosierende Veränderungen. Patienten mit ausgedehnter systemischer Inflammation und lymphozytärer oder neutrophiler Alveolitis (BAL) sind häufiger und schwerer betroffen. (4)
Im HR-CT stellt sich häufiger das Muster einer NSIP- als einer UIP-ähnlichen Fibrosierung dar. (5) CT-radiologische Veränderungen korrelieren zu funktionellen Einschränkungen. (6) Dabei kann die Beurteilung des HR-CT durch den visuellen Eindruck durch eine computergestützte Densitometrie verbessert werden. (7)
Eine umschriebene Gruppe von Patienten innerhalb des Spektrums der Sklerodermie weist klinische Besonderheiten auf. Diese Patienten haben meist eine umschrieben kutane Manifestation.
Zusätzlich kommen kutane, meist gelenknahne Kazifikationen vor. Kalzifikationen können insbesondere an den Unterschenkeln oder gelenknah an den Fingern zu schmerzlosen, areaktiv imponierenden Ulzerationen führen, die therapeutische schlecht beeinflußbar sind.
Schon Jahre vor dem klinischen Auftreten anderer Krankheitssymptome treten periphere Gefäßspasmen auf, die als Raynaud-Syndrom bezeichnet werden. Akrale Nekrosen, wenig mitfühlend auch „Rattenbiß-Nekrosen“ genannt sind typisch.
Oesophagus-Motilitätsstörungen können zu Refluxbeschwerden, Schluckstörungen und rezidivierenden Aspirationen führen. Im Thorax-CT stellt sich oft eine Dilatation des Oesophagus dar.
Eine Beteiligung der Finger mit derben, plumpen Auftreibungen wird Sclerodactylie genannt.
Teleangiektasien sind bevorzugt auf den Palmarflächen und auf den Wangen zu finden. (5,8)
CREST-Syndrom (5,8)
Calcinosis cutis
Raynaud-Syndrom
Esophagus-Motilitätsstörungen
Sklerodactylie
Teleangiektasien
Bei CREST-Syndrom sind weiterhin gehäuft Perikardergüsse zu beobachten und wird – da die kutane Manifestation ganz in den Hintergrund treten kann – auch als Systemic Sclerosis Sine Scleroderma bezeichnet. (5)
Die klinische Präsentation korreliert mit serologischen Markern. ANA sind in der Regel positiv. (5) Anti-SCL-70 und Anti-Topoisomerase I-AK sind mit einer hohen Frequenz einer Lungenbeteiligung verbunden. (8,9) Weitere diagnostische Marker für eine Lungenbeteiligung bei Sklerodermie sind Surfactant-Protein A und D. (10) Anti-Zentromer-Antikörer (anti-Th/To) sind typisch für das CREST-Syndrom. (5,8) Bei Patienten mit Anti-Zentromer-AK sind typischerweise keine Anti-SCL-70-AK vorhanden. (8,11)
Histologische stellt sich die Lungenbeteiligung bei Sklerodermie überwiegend als NSIP-Muster dar, seltener als UIP-Muster. Damit entspricht der histologische weitgehend dem radiologischen Befund. (12)
Funktionell imponiert zunächst eine diskrete Restriktion und leichte Einschränkung der Diffusion. (13) Zur Schweregradabschätzung ist eine Belastungsuntersuchung hilfreich. Eine einfache und gut reproduzierbare Belastungsuntersuchung, die für Sklerodermie-Patienten evaluiert ist, stellt der 6-Minuten-Gehtest dar. (14) Dabei ist die funktionelle Evaluation zur Prognoseeinschätzung geeigneter als die radiologische oder histologische Klassifizierung. (12)
Prognostisch wichtig ist die Entwicklung einer pulmonalen Hypertonie. In ca. 33 % der Patienten mit progressiver systemischer Sklerose und 50 % der Patienten mit CREST-Syndrom tritt eine pulmonale Hypertonie auf. (15) Risikofaktoren für eine pulmonale Hypertonie sind eine gleichzeitige fibrosierende Lungenerkrankung und ein fortgeschrittenes Alter. (16, 17) Typischerweise ist die pulmonale Hypertonie eine Komplikation nach etwa 20-jährigem Krankheitsverlauf und ist dann mit einer schlechten Prognose von etwa 2.5 Jahren verbunden. (18)
Patienten mit PH präsentieren oft akrale Nekrosen und Ulzera, entsprechend den Symptomen eines CREST-Syndroms. BSG und Gesamt-IgG sind erhöht, was für eine systemische Inflammation spricht. (19)
Radiologisch stellt sich eine erweiterte Pulmonalarterie dar und eine verbreiterte präkordiale Kontaktfläche zur Thoraxwand. (11) Im EKG spricht ein P-pulmonale (20) und schaufelförmige präkordial ST-Senkungen (11) für eine pulmonale Hypertonie.
Echokardiographisch läßt sich die Schwere der PH anhand des Flusses über die Trikuspidalklappe bestimmen. (11) In der Spiroergometrie ist schon eine belastungsinduzierte PH anhand eingeschränkter maximaler Sauersotffaufnahme und verminderter Belastunsgszeit faßbar. (21) Die Höhe der PH korreliert zu der Höhe des CO2-Atemäquivalents (22) und des alveolären PCO2. (23)
Grafik: PET CO2 und PPH (23) Grafik: Verlauf PET CO2 bei PPH unter Belastung (23)
Risikofaktoren für einen Progress einer leichten zu einer schweren PH sind höheres Lebensalter, limitierte kutane Erkrankung und bei Erstpräsentation erhöhter Pulmonalisdruck. (24)
Bislang existiert wenige evaluierte wirksame Therapien der Sklerodermie. Angewendet wurden unter anderem Steroide, Methotrexat, Cyclophosphamid, Chinin und Penicilamine ohne nachweisbaren signifikanten Effekt auf die Krankheitsprogression. (25) Ein Raynaud-Syndrom ist mit Calciumantagonisten beeinflußbar. (11)
Einzig eine längerfristige Therapie mit Cyclophosphamid konnte eine Besserung von Lungenfunktion, Dyspnoe, Belastbarkeit und von kutanen Beschwerden erbringen. (26) Dabei war lediglich der Effekt auf die Dyspnoe anhaltend nach Therapieende. (27) Eine Neutrophilie oder Eosinophilie in der BAL beschreibt zwar ein fortgeschrittenes Krankheitsbild, gibt aber keinen Hinweis auf die Wahrscheinlichkeit eines Therapieansprechens. (28)
Randomisierte Therapiestudien zur Therapie der PH bei Sklerodermie liegen nicht vor. Untergruppenanalysen von weiter gefaßten Studien ergaben keinen Unterschied im Therapieansprechen im Vergleich zur primären pulmonalen Hypertonie.
Daher wird empfohlen, nach gleichen Therapieprinzipien zu verfahren. (11) Zum Einsatz kommen u.a. Epoprostenol, Iloprost und Bosentan. (11, 29, 30). Der therapeutische Nutzen von Mycopheonal Mofetil ist nicht geklärt. (31)
Aufgrund der schlechten Prognose der PH bei Sklerodermie wird ein jährliches Screening mittels Echokardiographie empfohlen. (11, 32)
Bei dem berichteten Patienten mußte also die Diagnose eines langjährigen CREST-Syndromes mit pulmonaler Beteiligung und beginnender, noch leicht- bis mittelgradiger pulmonaler Hypertonie gestellt werden.
Da ein Therapieversuch mit Cylophosmphamid in der Vorgeschichte bereits schlecht vertragen worden war und zurzeit keine relevante pumlonale Einschränkung vorlag, wurde auf eine Wiederholung des medikamentösen Therapieversuchs verzichtet.
Eine Behandlungsindikation wegen der pulmonalen Hypertonie bestand noch nicht. Jedoch sind regelmäßige kurzfristige Kontrollen erforderlich, um ggf. eine medikamentöse Therapie einleiten zu können.
(1) Varga, J. Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin. Invest. 2007; 117: 557-567. Free Full Text
(2) Mayes, M.D., et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003 ; 48:2246-2255. Abstract
(3) Aguayo SM, Richardson CL, and Roman J. Severe extrapulmonary thoracic restriction caused by morphea, a form of localized scleroderma. Chest, Oct 1993; 104: 1304 - 1305. PDF
(4) Konig G, et al. Lung involvement in scleroderma. Chest, Mar 1984; 85: 318 - 324. PDF
(5) Fischer A, et al. Unique Characteristics of Systemic Sclerosis Sine Scleroderma-Associated Interstitial Lung Disease. Chest, Oct 2006; 130: 976 - 981. PDF Free Full Text
(6) Diot, E, Boissinot, E, Asquier, E, et al. Relationship between abnormalities on high resolution computed tomography and pulmonary function in systemic sclerosis. Chest, 1998:114,1623-1629. Abstract
(7) Camiciottoli G, et al. Lung CT Densitometry in Systemic Sclerosis: Correlation With Lung Function, Exercise Testing, and Quality of Life. Chest, March 1, 2007; 131(3): 672 - 681. Abstract
(8) Catoggio, LJ, Bernstein, RM, Black, CM, et al. Serological markers in progressive systemic sclerosis: clinical correlations. Ann Rheum Dis (1998) 42,23-37. PDF Abstract
(9) Diot E, et al. Is Anti-Topoisomerase I a Serum Marker of Pulmonary Involvement in Systemic Sclerosis? Chest, Sep 1999; 116: 715 - 720. PDF Free Full Text
(10) Takahashi H, et al. Serum Levels of Surfactant Proteins A and D Are Useful Biomarkers for Interstitial Lung Disease in Patients with Progressive Systemic Sclerosis. Am. J. Respir. Crit. Care Med. 162: 258-263. Abstract
(11) Olschewski H. Das CREST-Syndrom. In: Kirsten D, Costabel U (Hrsg.) Seltene Lungenkrankheiten 2. Inter-Pneu-Verlag, Großhansdorf. 2003. S 75ff.
(12) Bouros D, et al. Histopathologic Subsets of Fibrosing Alveolitis in Patients with Systemic Sclerosis and Their Relationship to Outcome. Am. J. Respir. Crit. Care Med. 165: 1581-1586, doi:10.1164/rccm.2106012. Abstract
(13) Owens GR, et al. Pulmonary function in progressive systemic sclerosis. Comparison of CREST syndrome variant with diffuse scleroderma. Chest, Nov 1983; 84: 546 - 550. PDF
(14) Villalba WO, et al. Six-Minute Walk Test for the Evaluation of Pulmonary Disease Severity in Scleroderma Patients. Chest, Jan 2007; 131: 217 - 222. PDF
(15) Ungerer RG, et al. Prevalence and clinical correlates of pulmonary arterial hypertension in progressive systemic sclerosis. Am J Med, Jul 1983; 75: 65-74. Abstract
(16) Schachna L, et al. Age and risk of pulmonary arterial hypertension in scleroderma. Chest, Dec 2003; 124: 2098-104. Free Full Text
(17) Chang B, et al. Scleroderma patients with combined pulmonary hypertension and interstitial lung disease. J Rheumatol, Nov 2003; 30: 2398-405. Abstract
(18) Cox SR, et al. Isolated pulmonary hypertension in scleroderma. Intern Med J, Jan 2005; 35: 28-33. Abstract
(19) Yamane K, et al. Clinical and laboratory features of scleroderma patients with pulmonary hypertension. Rheumatology, Nov 2000; 39: 1269-71. Free Full Text
(20) Wokhlu N, et al. P-wave amplitude and pulmonary artery pressure in scleroderma. J Electrocardiol, Oct 2006; 39: 385-8. Abstract
(21) Alkotob ML, et al. Reduced Exercise Capacity and Stress-Induced Pulmonary Hypertension in Patients With Scleroderma. Chest, Jul 2006; 130: 176 - 181. PDF
(22) Sun, XG, Hansen, JE, Oudiz, RJ, et al Exercise pathophysiology in patients with primary pulmonary hypertension. Circulation 2001;104,429-435. Free Full Text
(23) Yasunobu Y, Oudiz RJ, Sun XG, Hansen JE, and Wasserman K. End-tidal PCO2 Abnormality and Exercise Limitation in Patients With Primary Pulmonary Hypertension. Chest, May 1, 2005; 127(5): 1637 - 1646. Free Full Text
(24) Chang B, et al. Natural history of mild-moderate pulmonary hypertension and the risk factors for severe pulmonary hypertension in scleroderma. J Rheumatol, Feb 2006; 33: 269-74. Abstract
(25) Leighton C. Drug treatment of scleroderma. Drugs, Jan 2001; 61: 419-27. Abstract
(26) Tashkin DP, et al. Group. et al Cyclophosphamide vs placebo in scleroderma lung disease. N Engl J Med 2006;354,2655-2666. Abstract Free Full Text
(27) Tashkin DP, et al. Effects of 1-Year Treatment with Cyclophosphamide on Outcomes at 2 Years in Scleroderma Lung Disease. Am. J. Respir. Crit. Care Med. 176: 1026-1034. First published online as doi:10.1164/rccm.200702-326OC. Abstract
(28) Strange Ch, et al. Bronchoalveolar Lavage and Response to Cyclophosphamide in Scleroderma Interstitial Lung Disease. Am. J. Respir. Crit. Care Med. 177: 91-98. First published online as doi:10.1164/rccm.200705-655OC.Abstract
(29) Badesch DB, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med, Mar 2000; 132: 425-34. Abstract
(30) Girgis RE, et al. Long-term outcome of bosentan treatment in idiopathic pulmonary arterial hypertension and pulmonary arterial hypertension associated with the scleroderma spectrum of diseases. J Heart Lung Transplant, Oct 2005; 24: 1626-31. Abstract
(31) Gerbino AJ, et al. Effect of Mycophenolate Mofetil on Pulmonary Function in Scleroderma-Associated Interstitial Lung Disease
Chest, Prepublished online December 10, 2007 DOI:10.1378/chest.06-2861. Abstract
(32) Proudman SM, et al. Pulmonary arterial hypertension in systemic sclerosis: the need for early detection and treatment. Internal Medicine Journal (2007) 37:7, 485–494. Abstract
Dr. Holger Klee, 2 / 2008
zum Inhaltsverzeichnis zur Start-Seite
Reexpansionsödem nach Pneumothorax
zum Inhaltsverzeichnis zur Start-Seite
Ein 27-jähriger Mann berichtete über Belastungsdyspnoe rechts-thorakale Schmerzen, die seit 6 Tagen bestanden. Perkuttatorisch fand sich rechts hypersonorer Klopfschall und auskultatorisch kein Atemgeräusch.
Radiologisch stellte sich ein Pneumothorax mit Totalkollaps der rechten Lunge dar. Es wurde eine Bülaudrainage eingebracht und ein Sog von 20 cm Wassersäule angelegt. 10 Minuten später beklagte der Patient ziehende Schmerzen rechts und weiterhin Dyspnoe.
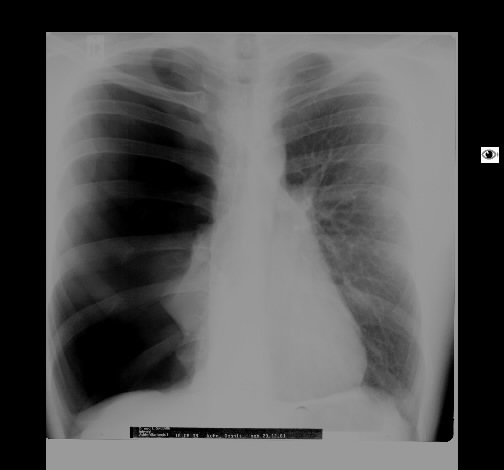
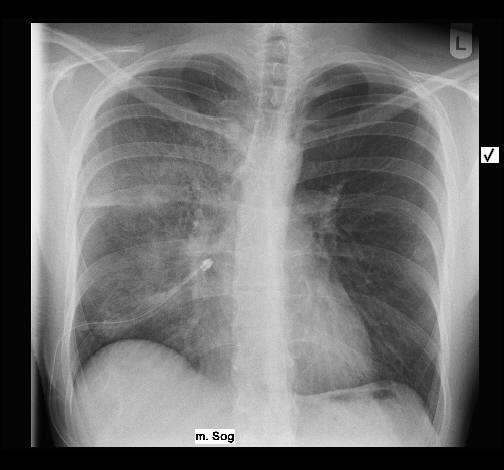
Rö-Thorax: Pneumothorax rechts Rö-Thorax: Drainage und Lungenödem rechts
Nach Reexpansion der rechten Lunge war ein interstitielles Lungenödem aufgetreten.
Reexpansionsödeme nach Drainage von Pleuraergüssen oder Pneumothoraces sind als Fallberichte schon seit ca. 150 Jahren vereinzelt beschrieben. (1) Eine Übersicht mit 146 Patienten mit Pneumothorax beschreibt eine Assoziation zu mittlerem Lebensalter (20 bis 39 Jahre) und zu einer größeren Ausmaß des Pneumothorax. (2) In einer größeren Arbeit über thorakoskopische Talkum-Pleurodese bei 460 Patienten wird eine Häufigkeit des Reexpansionsödem von 2.2 % angegeben. (3) Die Mortalität kann bis zu 20 % betragen. (4)
In einem Zeitraum von 10 Minuten bis zu 24 Stunden nach Drainage treten Reizhusten, Dyspnoe und evtl. Thoraxschmerzen auf. (1,4) Das Ödem kann sich auch auf nur einen Lappen beschränken. (5) Bei ausgedehntem Ödem kann auch eine lebensbedrohliche Kreislaufinstabilität und bei längerem Verlauf eine Pneumonitis auftreten. (6,7) Selten kann auch ein beidseitiges Lungenödem auftreten, evtl. als Folge einer Zytokinfreisetzung. (8)
Pathophysiologisch wurden unterschiedliche Mechanismen diskutiert. Ob durch negativen pleuralen Druck eine Transsudation in den Extrazuellularraum erfolgt ist fraglich. Die Ödem-Serum-Albumin-Ratio wurde mit 0.85 bestimmt. (9)
Der plötzlich erhöhte Sauerstoffpartialdruck führt zu einem Anstieg hochtoxischer freier Sauerstoffradikale, die ein pulmonares capillary leak syndrome auslösen. Es kommt zu einer Freisetzung verschiedener Zytokine, wie Interleukin-8, einem Neutrophilen-Chemotaktikum, und Leukotrien-B4. (10,11) Später kann pathologisch neben einem interstitiellen Ödem eine neutrophile Pneumonitis nachgewiesen werden. (7)
Die möglichweiser schwere Hypoxämie ist Folge eines Rechts-Links-Shunts. (2,6,7)
Tritt eine Kreislaufdepression auf, so kann als dies Folge einer Flüssigkeitssequestrations in die zuvor nur spärlich durchblutete Lunge gesehen werden. Es ist eine Hämokonzentration mit Anstieg des Hämatokrits geobachtet werden. Eine vorbestehende Hpoxämie kann zu dieser vorbestehenden Volumendepletion beitragen. Weiterhin kann nach Drainagebeginn eine myokardiale Depression als low-output failure auftreten. (6,7)
Die Wertigkeit der klassischen Risikofaktoren wurde in neueren Arbeiten hinterfragt.
Risikofaktoren des Reexpansionsödems
Atelektase länger als 3 Tage
?
„schnelle“ Drainage
??
Sog > 20 cm Wassersäule
??
Drainagemenge > 1 Liter
???
Die überwiegende Mehrzahl der Fallberichte schließen Patienten mit länger bestehendem Pneumothorax oder Pleuraerguß ein, so dass dies allgmein als Risikofaktor angesehen wird. (12) Ein besonderer Fallbericht unterstützt diese Annahme. Nach Drainage eines seit drei Tagen bestehenden Pneumothorax trat ein Reexpansionsödem auf, welches spontan regredient war. Danach kam es zu einem Rezidiv des Pneumothorax, welcher innerhalb von 17 Stunden komplikationslos drainiert wurde. (1) Hieraus kann durchaus schlüssig eine Abhängigkeit von der Dauer der Atelektase abgeleitet werden.
In einer experimentellen Arbeit mit Kaninchen, die eine Thorakoskopie simulieren sollte, konnte nach 60-Minuten einseitiger Ventilations- und Perfusionsunterbrechung zwar eine erhöhte Freisetzung von TNF-Alpha und IL-1ß, aber keine Veränderung der Permeabilität der pulmonalen Kapillaren nachgewiesen werden. (13) Daher ist anzunehmen, dass eine kurzfristige Ventilations-/Perfusionsunterbrechung, z.B. einer Thorakoskopie in Hinblick auf eine Reexpansionsödem weitgehend unproblematisch ist. Ein Reexpansionsödem nach Thorakoskopie ist eine sehr seltene Komplikation. (14)
Als „Expertenrat“ wird stets angeführt, dass eine „zu schnelle“ Drainage das Auftreten eines Reexpansionsödems begünstigen soll (12), wobei die Geschwindigkeit der Drainage nie näher quantifiziert wird. Ebenso wird empfohlen, den Sog möglichst niedrig zu wählen, jedenfalls nicht über 20 cm Wassersäule zu erhöhen. (12) Auch zur Unterstützung dieser Angabe gibt es keinerlei Studien. Selbst wenn eine Drainage ohne Sog, nur unter Anlage eines Wasserschlosses kann ein Reexpansionsödem auftreten. (2) Eine experimetelle Studie mit Reexpansionsödem bei Affen konnte eine eindeutige Abhängigkeit vom Sog (ohne Sog vs. 10 cm Wassersäule) darstellen (15), so dass weiterhin die Beschränkung des Sogs auf 15 bzw. 20 cm Wassersäule empfohlen wird. (3,12)
Eine hohe Drainagemenge über 1 bis 1.5 Liter wird regelmäßig als Risikofaktor für das Auftreten eines Reexpansionsödems angeführt. (1,2,3,6,12) In einer Studie bei 185 Patienten, die 2007 publiziert wurde, ließ sich kein Zusammenhang zwischen der Drainagemenge (bis 7.5 Liter) und dem Auftreten eines Reexpansionsödems nachweisen. (4) Daher empfehlen diese Autoren, stets eine vollständig Drainage von Ergüssen, solange der Sog nich 20 cm Wassersäule übersteigt und keine klinischen Beschwerden auftreten. (Ebenfalls nur „Expertenrat“)
Aus Einzelfallbeobachtungen wird abgeleitet, dass für das Auftreten eines kreislaufwirksamen Reexpansionsödems sind möglicherweise eine vorbestehende Hypoxämie, eine vorbestehende Hämokonzenteration und Hyponatriämie Risikofaktoren darstellen könnten. (6)
Das Reexpansionsödem ist oft oligosymptomatisch und bedarf dann keiner spezifischen Therapie. (2) Bei Hypoxämie infolge relevantem Shunt wird eine Sauerstoffgaben, ggf. auch Beatmungtherapie erforderlich. (2,14) Bei kreislaufstabilen symptomatischen Patienten Diuretika empfohlen. Tritt eine Kreislaufinstabilität ein, so ist ein ehebliches Volumendefizit anzunehmen. Dann ist eine Volumensubstitution bis zu 5 Litern und ggf. Katecholamingabe erforderlich. (6)
Da verschiedene Zytokine freigesetzt werden, von denen Interkeulin-8 wahrscheinilch das pathophysioloisch Relevanteste ist, wurde eine prophylaktische Gabe von Interkeulin-8-Antikörpergabe diskutiert. (13) In Anbetracht der Häufigkeit der Drainagetherapie (allein 1.3 Millionenen Thorakozentesen pro Jahr in den USA) und der relativen Seltenheit des Reexpansionsödems erscheint dies wenig kosteneffektiv. (3)
Bei dem obengeschilderten Patienten trat weder eine Hypoxämie noch eine Kreislaufinstabilität auf. Es erfolgte keine spezifische Therapie. Nach drei Tagen konnte die Drainagetherapie beendet werden.
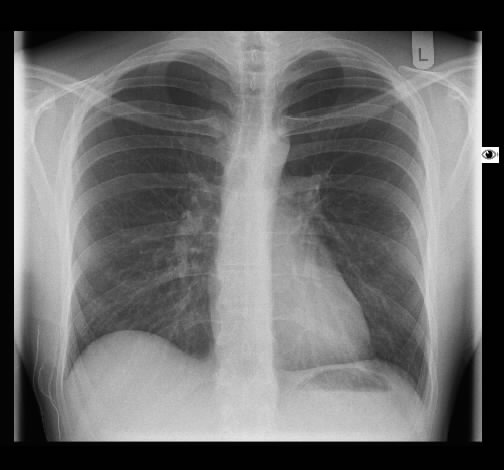
Rö-Thorax: restitutio ad integrum
(1) Mahajan VK, Simon M, and Huber GL. Reexpansion pulmonary edema
Chest, Feb 1979; 75: 192 - 194.
https://www.chestjournal.org/cgi/reprint/75/2/192
(2) Matsuura Y, etl a. Clinical analysis of reexpansion pulmonary edema. Chest, Dec 1991; 100: 1562 - 1566.
https://www.chestjournal.org/cgi/reprint/100/6/1562
(3) de Campos JRM, et al. Thoracoscopy Talc Poudrage : A 15-Year Experience
Chest, Mar 2001; 119: 801 - 806.
https://www.chestjournal.org/cgi/reprint/119/3/801
(4) Feller-Kopman D, et al. Large-Volume Thoracentesis and the Risk of Reexpansion Pulmonary Edema. Ann. Thorac. Surg., November 1, 2007; 84(5): 1656 - 1661.
https://ats.ctsnetjournals.org/cgi/reprint/84/5/1656
(5) VuongTK, et al. Reexpansion pulmonary edema localized to a lobe
Chest, May 1989; 95: 1170.
https://www.chestjournal.org/cgi/reprint/95/5/1170b
(6) Pavlin DJ, et al. Reexpansion hypotension. A complication of rapid evacuation of prolonged pneumothorax. Chest, Jan 1986; 89: 70 - 74.
https://www.chestjournal.org/cgi/reprint/89/1/70
(7) Sautter RD, et al. Fatal Pulmonary Edema and Pneumonitis After Reexpansion of Chronic Pneumothorax. Chest, Oct 1971; 60: 399 - 401.
https://www.chestjournal.org/cgi/reprint/60/4/399
(8) Ragozzino MW and Greene R, Bilateral reexpansion pulmonary edema following unilateral pleurocentesis. Chest, Feb 1991; 99: 506 - 508.
https://www.chestjournal.org/cgi/reprint/99/2/506
(9) Marland AM and Glauser FL. Hemodynamic and pulmonary edema protein measurements in a case of reexpansion pulmonary edema. Chest, Feb 1982; 81: 250 - 251.
https://www.chestjournal.org/cgi/reprint/81/2/250
(10) Nakamura H, et al. Elevated levels of interleukin-8 and leukotriene B4 in pulmonary edema fluid of a patient with reexpansion pulmonary edema. Am. J. Respir. Crit. Care Med. 149: 1037-1040.
https://ajrccm.atsjournals.org/cgi/content/abstract/149/4/1037
(11) Nakomura M, et al. Importance of Interleukin-8 in the Development of Reexpansion Lung Injury in Rabbits. Am. J. Respir. Crit. Care Med. 161: 1030-1036.
https://ajrccm.atsjournals.org/cgi/reprint/161/3/1030
(12) Thomsen TW, DeLaPena J, and Setnik GS. Thoracentesis. N. Engl. J. Med. 2006; 355(15): e16 - e16.
https://content.nejm.org/cgi/reprint/355/15/e16.pdf
(13) Funakoshi T, et al. Effect of re-expansion after short-period lung collapse on pulmonary capillary permeability and pro-inflammatory cytokine gene expression in isolated rabbit lungs. Br. J. Anaesth., April 1, 2004; 92(4): 558 - 563.
https://bja.oxfordjournals.org/cgi/reprint/92/4/558
(14) Barbetakis N, Samanidis G, Paliouras D, and Tsilikas C. Re-expansion pulmonary edema following video-assisted thoracic surgery for recurrent malignant pleural effusion. Interactive CardioVascular and Thoracic Surgery, June 1, 2008; 7(3): 532 - 534.
https://icvts.ctsnetjournals.org/cgi/reprint/7/3/532
(15) Miller MC, et al. Experimental pulmonary edema follwing reexpansion pneumothorax. Am Rev Respir Dis 1973;108:664.
Dr. Holger Klee, 9 / 2008
zum Inhaltsverzeichnis zur Start-Seite
Idiopathische Lungenhämosiderose (M. Ceelen)
zum Inhaltsverzeichnis zur Start-Seite
Ein 24-jähriger Patient stellte sich in der Notaufnahme vor wegen seit einigen Stunden anhaltenden mäßigen Hämoptysen. Es bestand keine Luftnot, kein Thoraxschmerz, kein Fieber, keine Allgemeinsymptomatik. Der Patient war zu vor stets gesund gewesen.
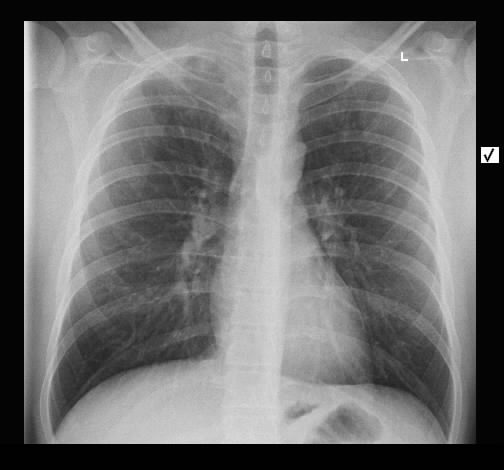
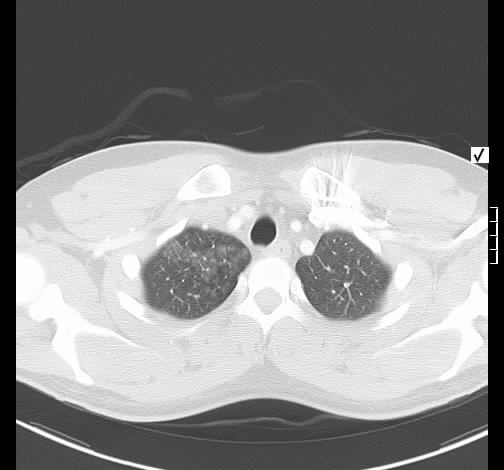
Rö-Thorax: flächige Trübung in rechten Oberlappen Thorax-CT: wolkige Infiltration rechts apikal
Das Routinelabor war einschließlich CRP vollständig unauffällig.
Labor (Norm)
Leuko
3.5 – 9.8
6.1
Hb
12.0 – 16.0
16.9
Thr
140 - 360
199
CRP
0.0 – 5.0
0.4
PCT
< 0.5
0.1
BSG
20 / 40
12 / 21
LDH
0 – 250
155
HST
10 – 50
27
Krea
0.4 – 1.2
1.2
Der Patient wurde am Aufnahmetag bronchoskopiert. Endoluminal stellte sich im rechten Oberlappen frisches Blut dar. Eine direkte Blutungsquelle fand sich jedoch nicht.
Gastroskopisch imponiete Hämatin und altes Blut im Magen, jedoch ebenfalls keine Blutungsquelle.
Eine HNO-ärztliche Endoskopie ergab einen Normalbefund.
Die Differentialdiagnostik der alveolären Hämorrhagie umfaßt insbesondere Vaskulitiden. Hämoptysen kommen auch bei infektiologischen und neoplastischen Krankheitsbildern vor.
Differentialdiagnose der alv. Hämorrhagie
Vaskulitiden
ANCA, ANA
Pneumonie, Bronchitis
Klinik, CRP, PTC
Tuberkulose
SFS, TIGRA
Goodpasture-Syndrom
Anti-BM-AK
Idiopath. Hämosiderose
Ausschluß, BAL
Differentialdiagnose der alv. Hämorrhagie (10)
Bei dem gesschilderten Patienten verlief die Differentialdiagnostik samätlich ergebnislos, so dass als Ausschlußdiagnose eine idiopathische Lungenhämosiderose (M. Ceelen) in Betracht gezogen wurde.
Die idiopathische Lungenhämosiderose ist klinisch durch Hämoptysen, pulmonale Infiltrate und möglicherweise eine mikrozytäre Anämie gekennzeichnet. (1,2) Die Ätiologie und Pathogenese der idiopathischen Lungenhämosiderose ist (wie der Name schon sagt) unklar. Zirkulierende Immunkomplexe und Immunkomplexablagerungen an der alveolären Basalmembran legen eine Autoimmungenese nahe. (3) Möglicherweise besteht eine Assoziation zu Kuhmilch-Sensibilisierung oder zur Zoeliakie. (3, 9)
Wenn sich im Verlauf eine benennbare Autoimmunerkrankung demaskiert (ANCA-assoziierte Vaskulitis, SLE oder andere), sollte nicht mehr von einer idiopathischen Lungenhämosiderose gesprochen werden. (4)
Das Krankheitsbild betrifft zu 80 % Kinder und Jugendliche. Im Erwachsenenalter manifestiert sich die Erkrankung typischerweise in der 4. bis 5. Dekade. (3) Eine Serie beschreibt eine familiäre Häufung (Großmutter, zwei Schwestern), so dass möglicherweise auch eine genetische Prädiposition vorliegn könnte. (5)
Wie oben angeführt, stehen rezidvierende Hämoptysen im Vordergrund, die ggf. zu einer mirkozytären Anämie führen können. (3)
Klinik des M. Ceelen (6) %
Anämie
100
Pulmonale Infiltrate
100
Husten
100
Hypoxämie
85
Fieber
70
Luftnot
60
Trommelschlegelfinger
17
Blässe
17
Der Krankheitsverlauf ist bei insbesondere bei Kindern in der Regel progredient und kann zu fibrotischen Lungeveränderungen und chronischen respriatorischem Versagen führen. (7) Ebenso kann eine Makrohämatoe letal verlaufen. (6)
Radiologisch imponieren wolkige Infiltrate, welche meist basal anzutrefen sind. Bei chronisch rezidivierendem Velauf kann ein fibrotischer Umbau auftreten.
In der Histologie und der BAL finden sich neben einer alveolären Hämorrhagie charakterischerweise Hämosiderin, welches in den Alveolarmakrophagen akkumuliert. (3) Die Bronchoalveoläre Lavage wird typischerweise im Verlauf der Untersuchung mit jeder Portion blutiger. (3)
Bild nicht vorhanden
Die immunsuppressive Therapie umfaßt in aller Regel eine Langzeittherapie mit systemischen Steroide (75%) und in der Hälfte der Fälle Hydrochloroquine oder Azathioprin. (6) Bei fulminanten Verläufen kann die Gabe von Cyclophsphamid in Betracht gezogen werden. (8)
Die Prognose wurde in den 60er-Jahren noch als schlecht eingeschätzt. Unter immunsuppressiver Therapie ist bei pädiatrischen Patienten mit einer 5-Jahres-Prognose von 86% zu rechnen (6). Wenn eine Assoziation zu einer Zöliakie besteht, kann evtl. unter ensprechenden diätetischen Maßnahmen die Prognose verbessert werden. (9)
Bei dem oben beschriebenen Patienten sistierten unter einer systemischen Steroidmedikation mit 50 mg Prednisolon-Äquivalent pro Tag nach zwei Tagen die Hämoptysen. Er befindet sich in regelmäßiger ambulanter Nachbetreuung und ist seit einem Jahr beschwerdefrei.
(1) Richard S. Irwin, Thomas S. Cottrell, Konrad C. Hsu, William R. Griswold and Henry M. Thomas III
Idiopathic Pulmonary Hemosiderosis: An Electron Microscopic and Immunofluorescent Study
Chest 65:41-45
https://www.chestjournal.org/content/65/1/41.full.pdf
(2) C J Dolan, Jr, C H Srodes and F D Duffy
Idiopathic pulmonary hemosiderosis. Electron microscopic, immunofluorescent, and iron kinetic studies.
Chest 68:577-580
https://www.chestjournal.org/content/68/4/577.full.pdf
(3) Octavian C. Ioachimescu, Carol Farver and James K. Stoller
Hemoptysis in a 77-Year-Old Male With a Systolic Murmur*
Chest 128:1022-1027
https://www.chestjournal.org/content/128/2/1022.full.pdf
(4) Richard B. Byrd and Gary Trunk
Systemic Lupus Erythematosus Presenting as Pulmonary Hemosiderosis
Chest 64:128-129
https://www.chestjournal.org/content/64/1/128.full.pdf
(5) R L Breckenridge, Jr and J S Ross
Idiopathic pulmonary hemosiderosis: a report of familial occurrence.
Chest 75:636-639
https://www.chestjournal.org/content/75/5/636.full.pdf
(6) Muhammad M. Saeed, Marlyn S. Woo, Eithne F. MacLaughlin, Monique F. Margetis and Thomas G. Keens
Prognosis in Pediatric Idiopathic Pulmonary Hemosiderosis*
Chest 116:721-725
https://www.chestjournal.org/content/116/3/721.full.pdf
(7) D L Buschman and R Ballard
Progressive massive fibrosis associated with idiopathic pulmonary hemosiderosis.
Chest 104:293-295
https://www.chestjournal.org/content/104/1/293.full.pdf
(8) J L Colombo and S M Stolz
Treatment of life-threatening primary pulmonary hemosiderosis with cyclophosphamide.
Chest 102:959-960
https://www.chestjournal.org/content/102/3/959.full.pdf
(9) A Pacheco, C Casanova, L Fogue and A Sueiro
Long-term clinical follow-up of adult idiopathic pulmonary hemosiderosis and celiac disease.
Chest 99:1525-1526
https://www.chestjournal.org/content/99/6/1525.full.pdf
(10) Brown KK, Pulmonary Vasculitis. The Proceeding of the American Thoracic Society 3:48-57 (2006)
Dr. Holger Klee, 3 / 2009
zum Inhaltsverzeichnis zur Start-Seite
zum Inhaltsverzeichnis zur Start-Seite
Ein 39-jähriger Patient beklagte Reizhusten und eine geringe Dyspnoe, wenn er schnell mehr als zwei Etagen steige. Fieber, purulentes Sputum, Thoraxschmerzen oder eine Allgemeinvesrchlechterung bestand nicht.
Seit der Kindheit sind häufig grippale Infekte und eine chronische Sinusitis bds. aufgetreten. Hin und wieder bestehen abdominelle Beschwerden, die von Durchfall begelitet werden. Vor einem halben Jahr war eine stationäre Behandlung wegen einer infektiösen Durchfallerkrankung notwendig gewesebn.
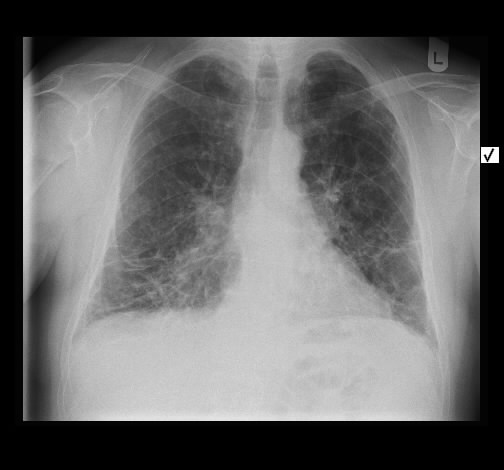
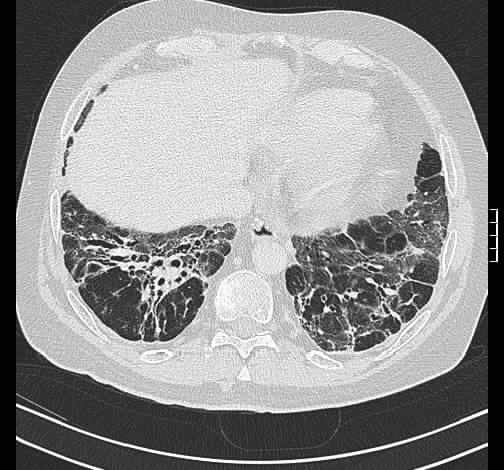
Rö-Thorax: unterlappen-betonte retikuläre Zeichungsvermehrung bds. Thorax-CT: retikuläre Zeichnungsvermehrung, UL-betont, Bronchiektasen
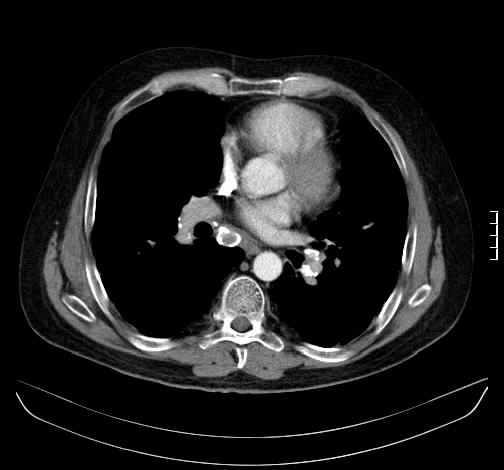
Thorax-CT: vergrößerte mediastinale LK mit schalenförmiger Verkalkung Rö-NNH: flächenförmige Weichteilschwellung, keine Spiegelbildung
Das Routinelabor war zunächst unauffällig, abgesehen von einer Thrombopenie. Keine erhöhten Enzündungsparameter, ANA, ANCA negativ. ACE und sIL3 waren im Normbereich. Die Elektrophorese zeigt einen Mangel an Gamma-Globulinen.
Die broncho-alveoläre Lavage ergab eine lymphozytäre Alveolitis.
Broncho-Alveoläre Lavage
(Norm)
Alveolarmakrophagen
43 %
86 – 98 %
Lymphozyten
52 %
0 – 10 %
Neutrophile Granulozyten
5 %
0 – 3 %
Eosinophile Granulozyten
0 %
0 – 1 %
CD 4 / CD 8-Quotient
2.5
2 - 3
Die Histologie einer peripheren Lungen-PE zeigte nicht-verkäsende epitheloidzellige Granulome.
Hautveränderung
Auf Nachfragen zeigte der Patient an den Unterschenkeln livide, erhabene Stellen, die nicht schmerzhaft seien und schon seit der Kindheit bestanden.
Lungenfunktion
Leichte Restriktion, leicht eingeschränkter Transferkoeffizient, normale Blutgase.
Lungenfunktion
VC
3.4
98 % des Solls
FEV-1
2.7 l
79 % der VC
TLC
5.1 l
76 % des Solls
R(tot)
0.2
TLCO/VA
73 % des Solls
pO2
82 mm Hg
pCO2
34 mmHg
Mittelgradig verminderte Leistungsfähigkeit bei Zeichen der Diffusionsstörung (AaDO2 52 mmHg) mit mäßiger belastungsinduzierbarer Hypoxämie (PaO2min. 63 mmHg). Relative Hyperventilation (VE/VCO2 50). Kein restriktives Atemmuster. Kein Hinweis auf kardio-zirkulatorische Limitierung.
Spiroergometrie
VO2max.
1.730 l/min
62 % des Solls
AaDO2max.
52 mmHg
erhöht
VE/VCO2
50
erhöht
PaO2min.
63 mmHg
erniedrigt
Differentialdiagnose
Die oben geschilderten pulmonalen Veränderungen sind sehr gut mit einer Sarkoidose vereinbar, wenn auch ACE und sIL2R nicht erhöht sind. Auffällig war jedoch der ausgeprägte Gamma-Globulinmangel. Daher ergibt sich in Zusammenschau mit den geschilderten rezidivierenden Infekten die Diagnose eines varibalen Immundefektes (Common variable Immunodeficiency CVID). Die granulomatösen Veränderungen treten auch im Rahmen dieser Grunderkrankung auf.
Epidemiologie und Pathogenese
CVID ist ein angeborenes Immundefektsyndrom mit einer Inzidenz von 2 – 12 / 100.000 Geburten. (1, 2) Es ist charakterisiert durch einen Mangel an Immunglobulinen, insbesondere IgG ohne geleichzeitigen zellulären Immundefekt. (1) Obwohl schon oft seit der Kindheit symptomatisch, wird die Diagnose im dritten bis vierten Lebensjahrzehnt gestellt. (1, 5)
Ca. 80 % der Patienten mit CVID weisen eine normale Anzahl peripherer B- und T-Zellen auf. Die in vitro-stimulierbare Produktion von Ig-Isotypen kann vermindert, oder auch normal sein trotz Gesamt-IgG in vivo. Eine Minderzahl der Patienten (5-10 %) weisen einen peripheren B-Zell-Mangel auf. Eine weitere kleine Gruppe (5-10 %) zeigt nicht-verkäsende eptheloidzellige Granulome in verschiedensten Organen und neigt zusätzlich zu einem progressiven T-Zell-Defekt. (2)
Klinik
Die Klinik des CVID ist außerordentlich heterogen. Anhand der peripheren differenzierten B-Zellen (CD27+IgM-IgD-) ist eine klinische Einteilung vorgeschlagen worden. (2, 3, 4)
Sind differenzierte Memory B-Zellen in normaler Zahl vorhandeln (MB2), bestehen häufig rezidivierende Infekte der oberen und unteren Atemwege wie Sinusitiden und Pneumonien, sowie Durchfallerkrankungen. In der Gruppe mit verminderten Memmory B-Zellen (MB1) sind zusätzlich Splenomegalien anzutreffen. Sind keine Memory B-Zellen vorhanden (MB0), ist oft mit Splenomegalie, lymphoider Proliferation und granulomatösen Erkrankungen, sowie bei der Hälft der Patienten mit Durchfallerkrankungen und Malabsortionssyndrom (4) zu rechnen. In dieser Gruppe betreffen chronische Lungenerkrankung, wie Bronchiektasen, obstruktive und restriktive Lungenfunktionsstörungen die Hälfte der Patienten. (4)
Klinik des CVID
Rezidivierende Atemwegsinfekte, Bronchiektasen
Durchfallerkrankungen, Malabsortion
Autoimmunerkrankungen
Autoimmunerkrankungen, wie Immunthrombopenien und Autoimmunhepatitiden kommen in allen Gruppen vor und betreffen ca. 20 % der Patienten. (3)
Autoimmunerkrankungen bei CVID
Immun-Thrombopenie
30 %
Hämolytische Anämie
20 %
Rheumatoide Arthritis
20 %
SLE, PBC, periniziöse Anämie, Thyreoitis
Patienten mit CVID haben im Vergleich zur Normalbevölkerung ein 8-fach erhöhtes Risiko, eine Neoplasie zu entwickeln. Der Gipfel der Manifestationsalter liegt in der 5. bis 6. Dekade.
Neoplasien bei CVID
Non-Hodgkin-Lymphome
50 %
Adeno-Magencarcinome
28 %
Colon-Carcinome
18 %
Lungenerkrankungen bei CVID
Im Vordergrund stehen rezidivierende pulmonale Infekte mit einem Risiko der Bronchiektasenentwicklung. (5, 6) Obstruktive Ventilationsstörungen betreffen insbesonder Raucher. (9) Jedoch kann ein ganzes Spektrum an interstitiellen Lungenerkrankung bei CVID gesehen werden. Neben einer BOOP (7) wurden in einer Gruppe von 148 CVID-Patienten 20 Patienten mit ILD beschrieben, die klassifiziert wurden als neutrophile Alveolitiden, LIP, NSIP, UIP und Sarkodiose-ähnliche Erkrankungen. (8)
Granulomatöse Erkrankungen bei CVID
Eine Subgruppe von ca. 10 % der Patienten mit CVID weist nicht-verkäsende epitheloidzellige Granulome in unterschiedlichen Organsystemen auf. (2, 10, 11) Dies kann zu organspezifischen Dysfunktionen und Komplikationen führen. Veränderungen an Lunge und mediastinalen Lymphknoten sind nicht von einer Sarkoidose unterscheidbar. ACE ist auch bei CVID-Patienten erhöht. Dennoch ist für Sarkoidose eine Hypergammoglobulinämie typisch. So sollte eine Hypogammaglobulinämie bei vermuteter Sarkoidose die Differentialdiagnose eines CVID nach sich ziehen.
CVID-Patienten mit granulomatösen Veränderungen sind prädisponiert für Autoimmunerkrankungen (53 %) und T-Zell-Defizite. (11)
Therapie
Der zentrale Therapieschritt des CVID ist die Substitution von Immunglobulinen. Der therapeutische Zielbereich sind 5 g/l. Auch hierunter können nicht immer rezidivierende Infekte und ein Fortschreiten einer Bronchiektasenentwicklung vermieden werden. (10) Rezdivierende Sinusitiden sollten HNO-ärztlich betreut werden mit ensprechenden Lokalmaßnahmen, wie beispielsweise täglichen Nasenduschen.
Bei Autoimmunerkrankungen kommt ggf. systemische Steriodmedikation oder bei Thrombopenie eine Splenektomie in Betracht.
Fallbericht
Der Patient wurde mit Immuglobulinen substituiert und in HNO-ärztliche Betreuung entlassen. Die Häufigkeit der Atemwegsinfekte nahm deutlich ab.
Fünf Jahre nach Diagnosestellung trat eine Verschlechterung der pulmonalen Situation ein, so daß unter der Annahme einer Exazerbation der granulomatösen Veränderungen eine systemische Steroidtherapie begonnen wurde. Dies führte zu einer Stabiliserung, so dass die systemische Steroidmedikation reduziert werden konnte.
Literatur
(1) Ali El-Solh, Pawan Sikka, and Azmi Draw. A 58-Year Old Woman With Recurrent Productive Cough and Diarrhea, Chest October 2000 118:1194-1197.
(2) Warnatz, K, Denz, A, Drager, R, et al Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood 2002;99,1544-155
(3) Piqueras, B, Lavenu-Bombled, C, Galicier, L, et al Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J Clin Immunol 2003;23,385-400
https://www.ncbi.nlm.nih.gov/pubmed/14601647?dopt=Abstract
(4) Drahomíra Detková, Javier de Gracia, Susana Lopes-da-Silva, et al. Common Variable Immunodeficiency, Association Between Memory B Cells and Lung Diseases. Chest June 2007 131:1883-1889
(5) Mace SR, et al. Pulmonary Clinico-pathological findings in Common Variable Immunodeficienty. Interstitial Lung Disease. Chest October 1998 114:347S-350S
(6) Kainulainen, L, Varpula, M, Liippo, K, et al Pulmonary abnormalities in patients with primary hypogammaglobulinemia. J Allergy Clin Immunol 1999;104,1031-1036
https://www.ncbi.nlm.nih.gov/pubmed/10550749?dopt=Abstract
(7) J Kaufman and R Komorowski, Bronchiolitis obliterans organizing pneumonia in common variable immunodeficiency syndrome.Chest August 1991 100:552-553; doi:10.1378/chest.100.2.552
(8) Valentin Popa, Thomas V. Colby, and Stanley B. Reich Pulmonary Interstitial Disease in Ig Deficiency*Chest November 2002 122:1594-1603
(9) V Popa, Airway obstruction in adults with recurrent respiratory infections and IgG deficiency.Chest April 1994 105:1066-1072
(10) Fasano, MB, Sullivan, KE, Sarpong, SB, et al Sarcoidosis and common variable immunodeficiency: report of 8 cases and review of the literature. Medicine 1996;75,251-261
https://www.ncbi.nlm.nih.gov/pubmed/8862347?dopt=Abstract
(11) Mechanic, LJ, Dikman, S, Cunningham-Rundles, C Granulomatous disease in common variable immunodeficiency. Ann Intern Med 1997;127,613-617
Dr. Holger Klee, 3 / 2009
zum Inhaltsverzeichnis zur Start-Seite
{kind=link}